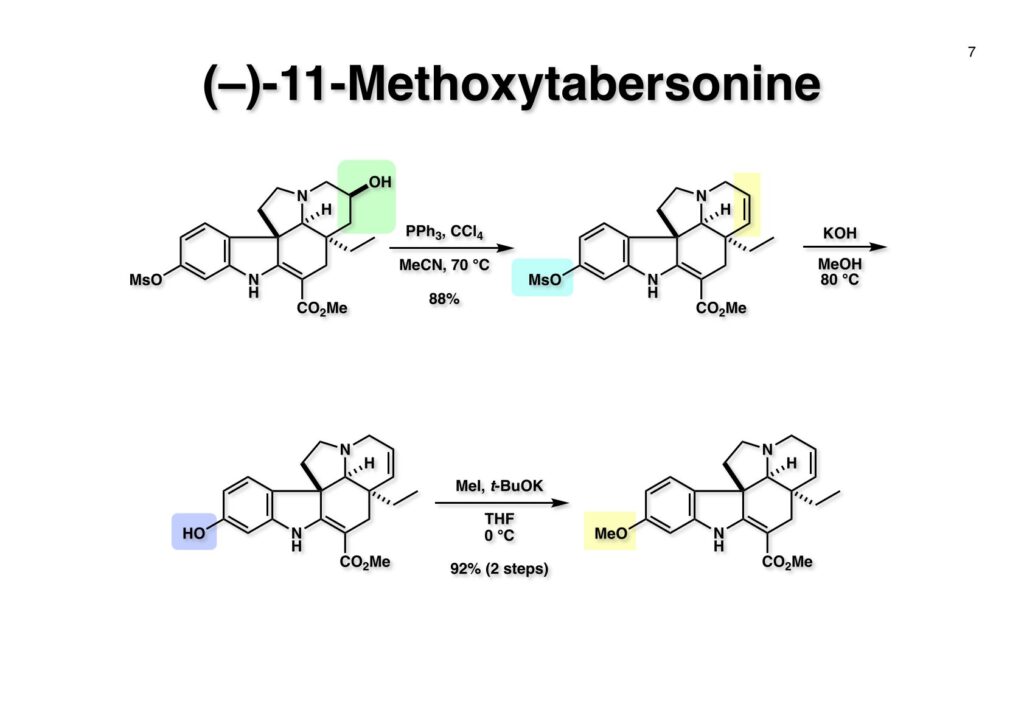
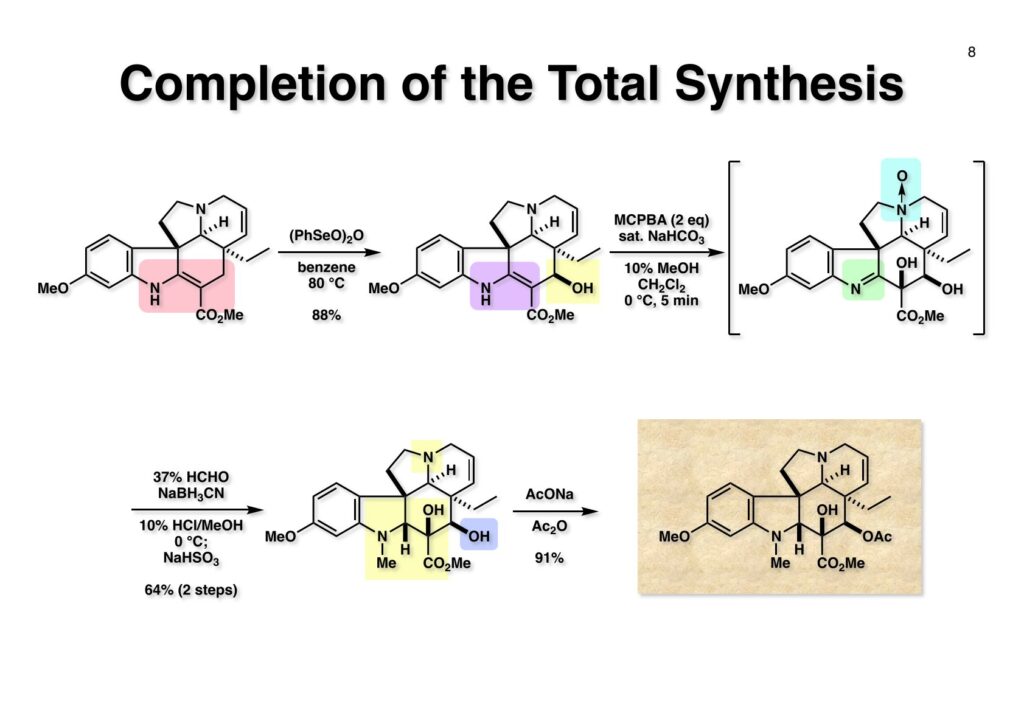
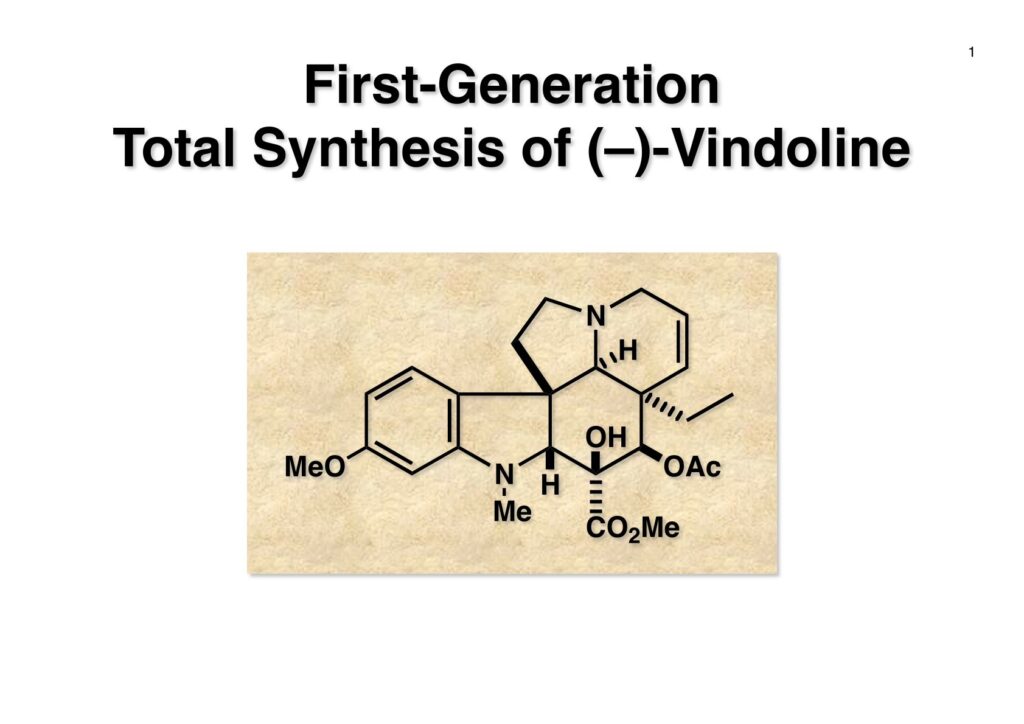
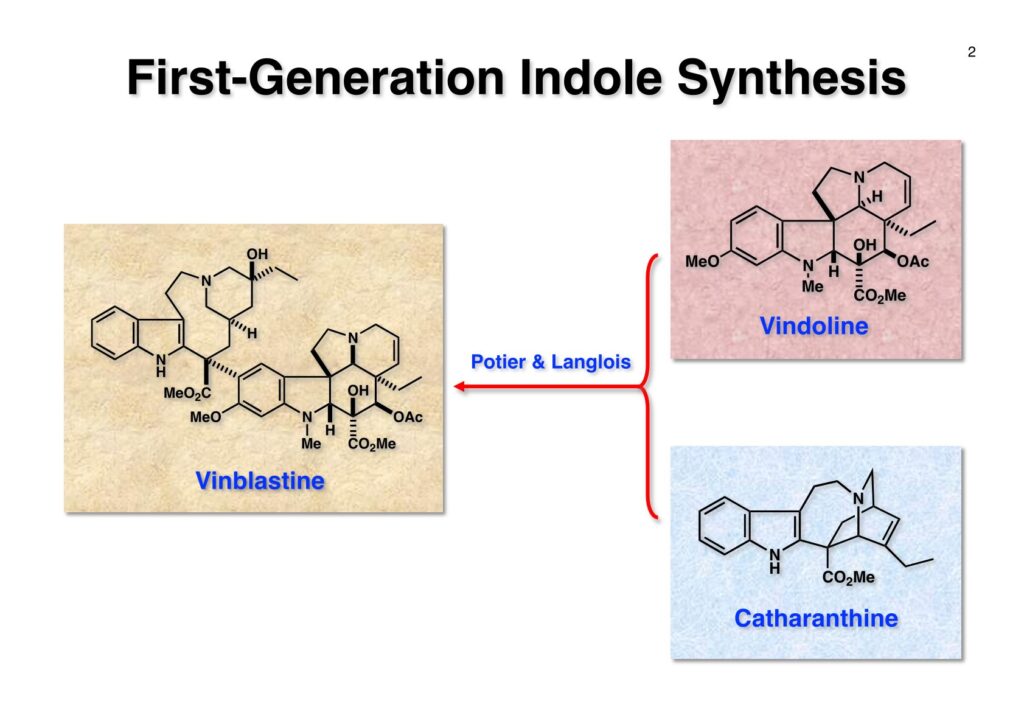
第一世代インドール合成法を使ってvincadifformineの全合成をライス大学で行ったが、東大に赴任してもっと複雑な構造を有するvindolineの全合成をやってみたくなった。丁度、三共株式会社から小林聡さんが研究生として来られたのでvindolineの合成に向けてまず光学活性なtabersonineの全合成を開始した。その後、日本新薬から研究生として来られた上田稔浩さんにも加わってもらいvindolineの全合成が完成した。しかし、このルートではvinblastineの全合成は遥か彼方の目標で、catharanthineの全合成で開発した第二世代インドール合成法の出現で高効率的なvindolineとそれに連なるvinblastineの全合成を成し遂げることができた。
“An Efficient Total Synthesis of (–)-Vindoline,” S. Kobayashi, T. Ueda, and T. Fukuyama,. Synlett, 883-886 (2000).
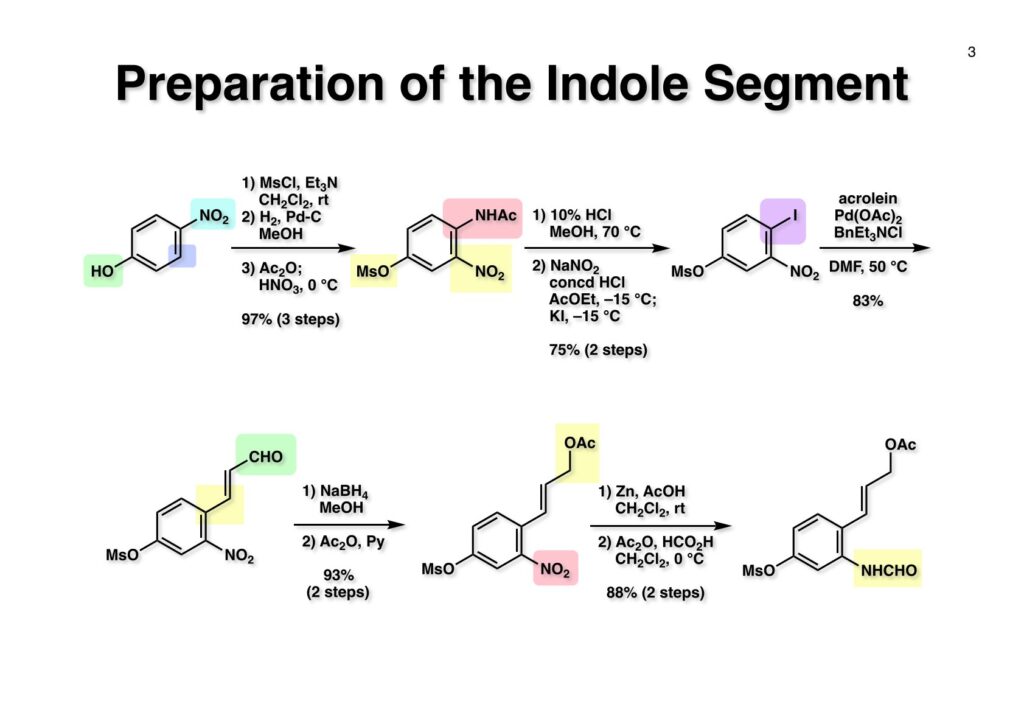
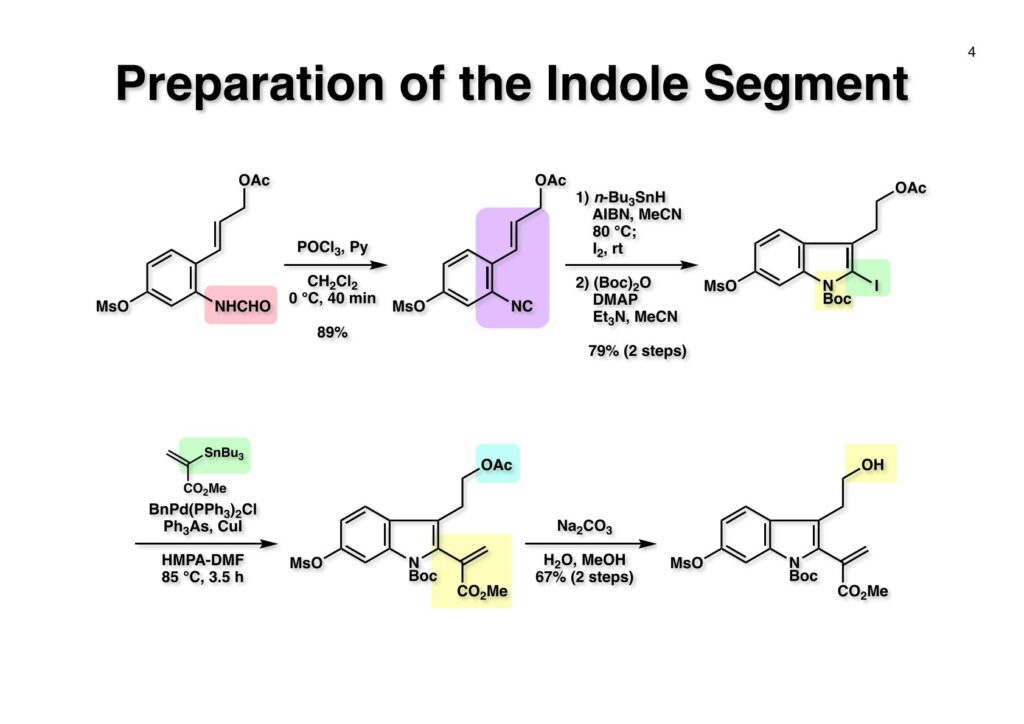
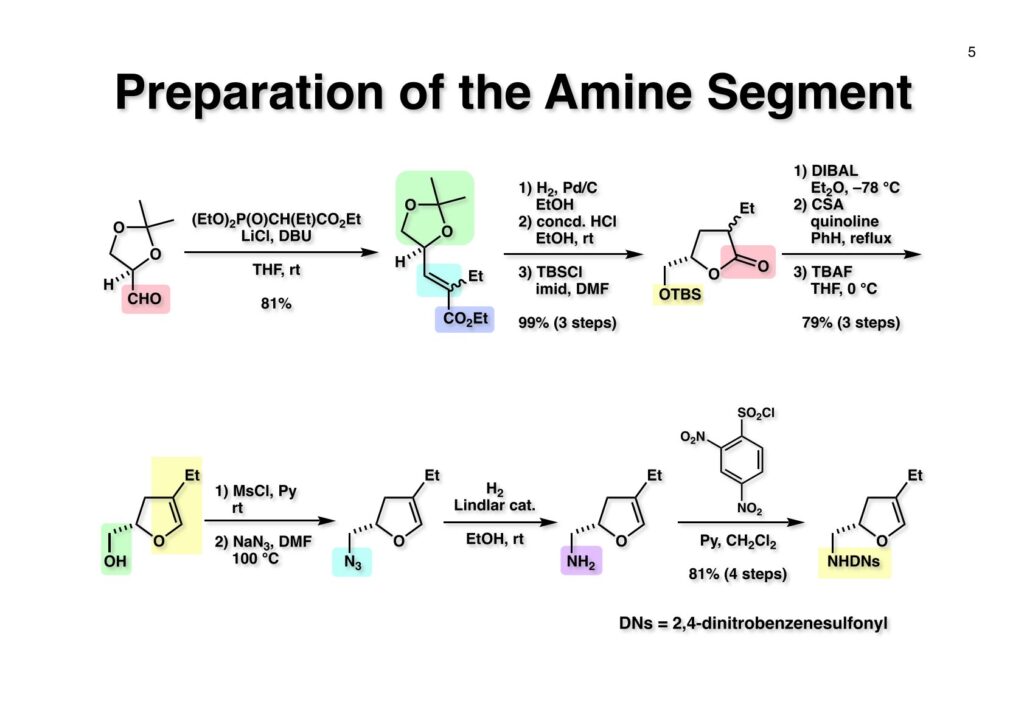
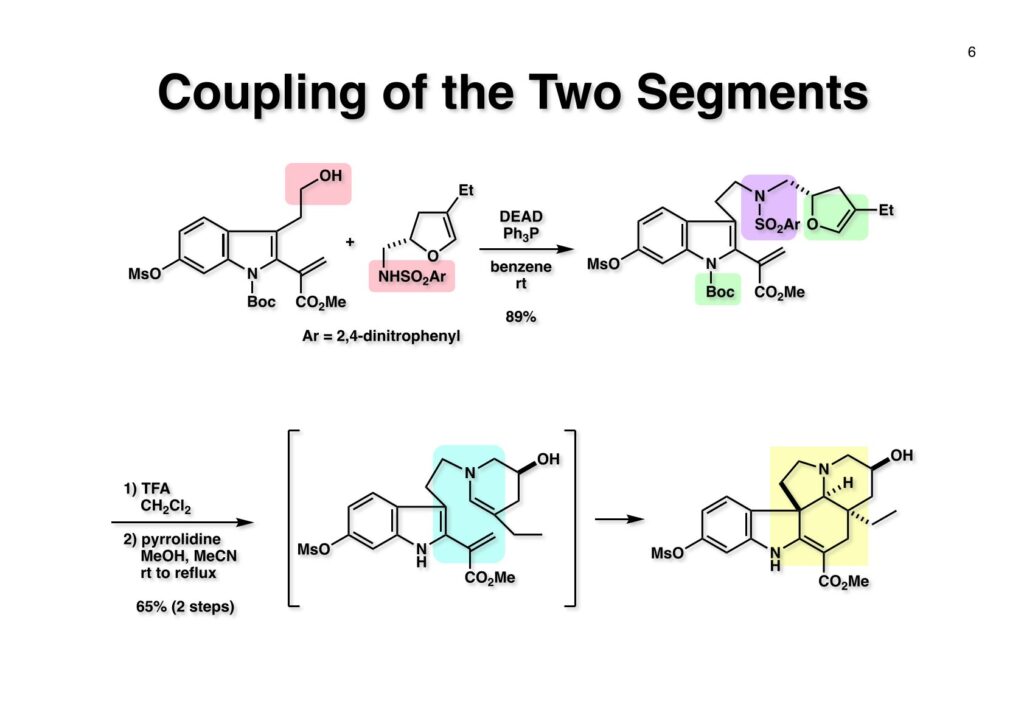
D-Mannitolから2段階で容易に得られるグリセルアルデヒドのアセトナイド (1-1) をRoush-正宗によるHorner-Emmons反応の改良法で (1-2) のジアステレオマー混合物を得た。(1-2) の二重結合を水添し、塩酸/EtOHでアセトナイドを除去すると、より安定な5員環ラクトンが生成し、水酸基をTBS化して (1-3) を高収率で得た。次にラクトンをDIBAL還元してラクトールに変換し、CSA-quinoline存在下ベンゼン中で加熱すると脱水してジヒドロフラン体が生成する。このTBS基をTBAFで除去して (2-1) が得られた。(2-1) は2段階でアジド (2-2) に変換し、二重結合を還元しないようにLindlar触媒を使ってアジドをアミン (2-3) に還元した。アミン (2-3) を2,4-dinitrobenzenesulfonyl chloride (2-4) と反応させてアミン部分 (2-5) が得られた。