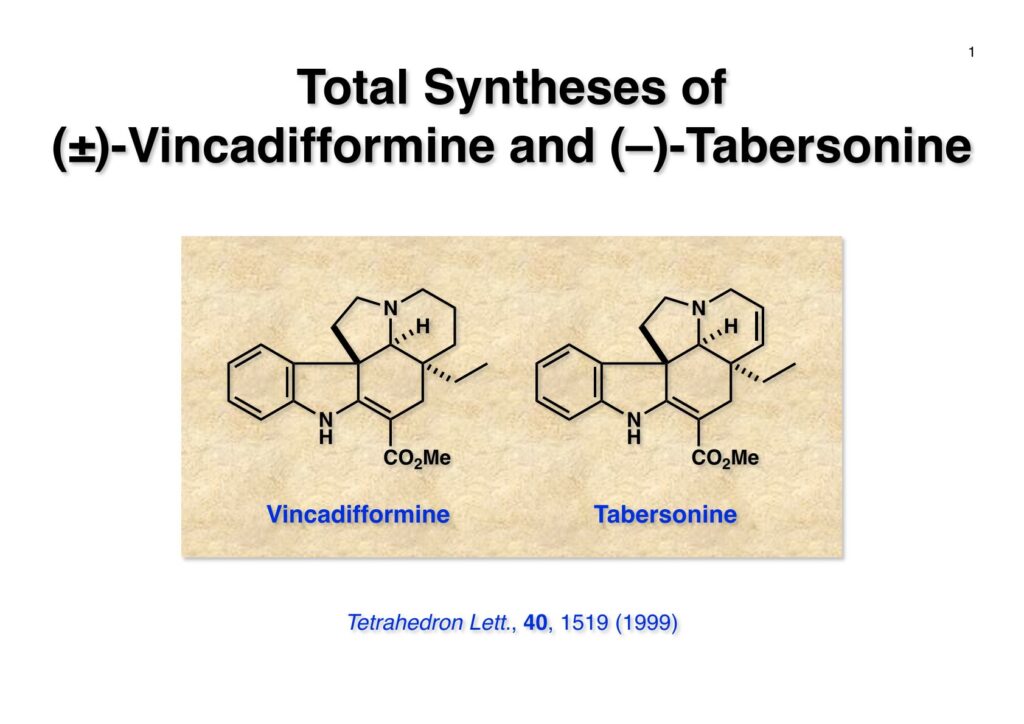
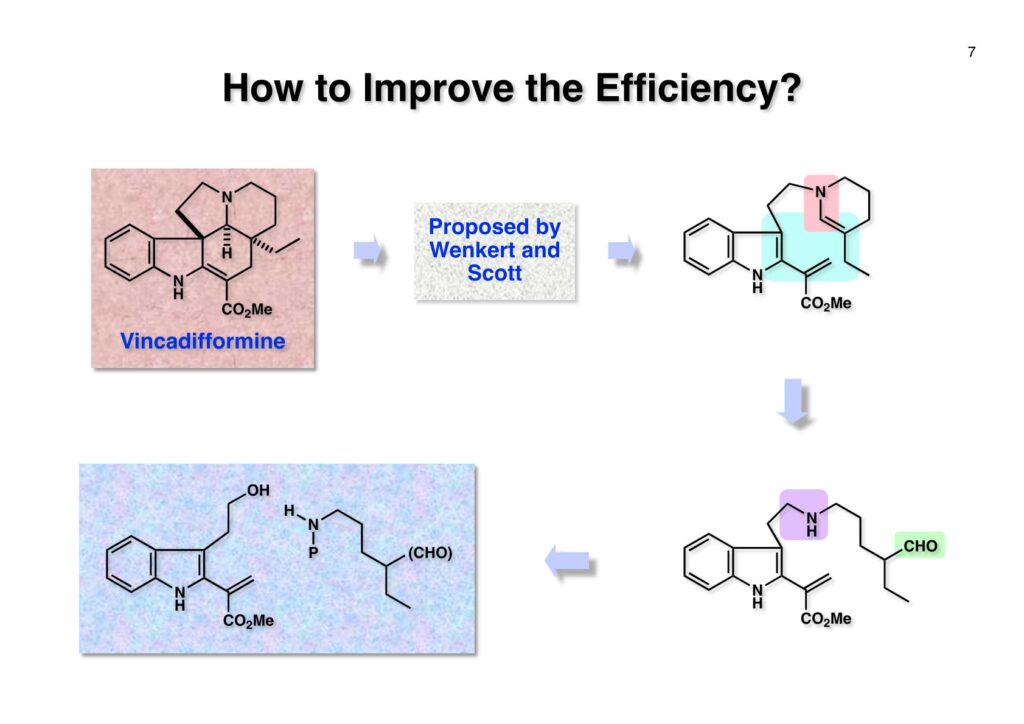
Vincadifformineは第一世代のインドール合成法を開発したので、手始めにインドールアルカロイドの全合成に適用してみようと思って始めたプロジェクトだ。南開大学卒のGe Pengという非常に真面目な女子院生にやってもらい1994年には全合成が完了した。彼女はサンフランシスコのNutrixというスタートアップ製薬会社で研究を続けている。日本に帰国後もMui Cheungの成果同様になかなか論文を書かなかったが、三共株式会社から小林聡さんが研究員として来られたので、vindolineの全合成のモデル実験としてのtabersonine全合成をやってもらい、抱き合わせで論文を出す事ができた。Vincadifformineの全合成は新規インドール合成法が活用されただけでなく、アミンの活性化のために2,4-dinitrobenzenesulfonamideを見つける端緒となった研究であり、極めて意義深い研究だったと言える。
“Efficient Total Synthesis of (±)-Vincadifformine and (–)-Tabersonine,” S. Kobayashi, G. Peng, and T. Fukuyama, Tetrahedron Lett. , 40 , 1519-1522 (1999).
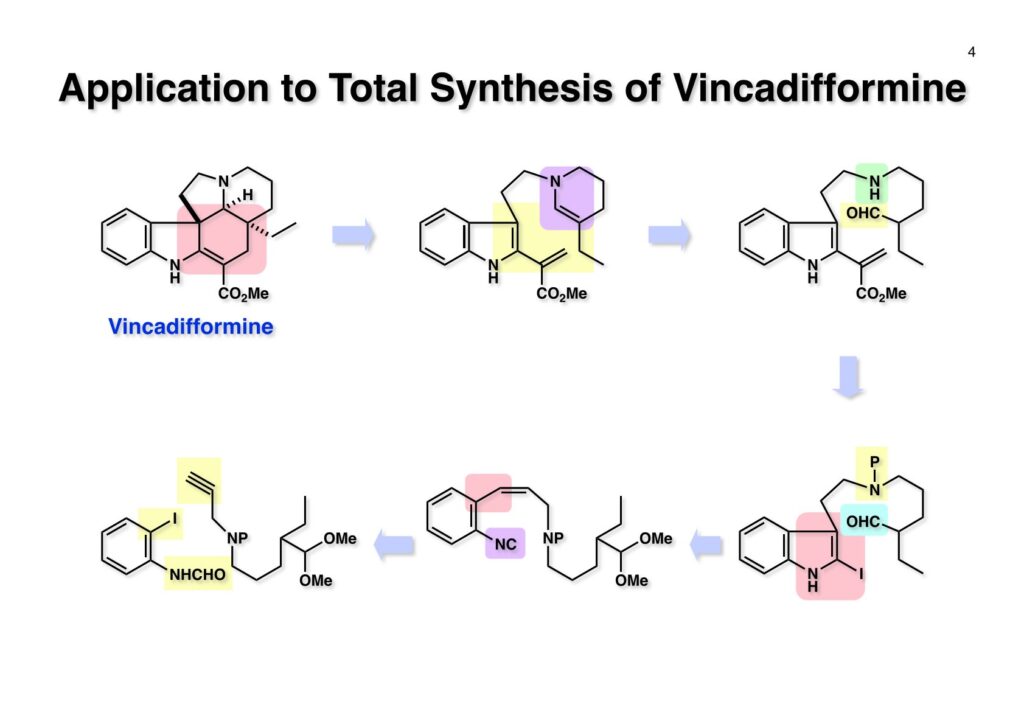
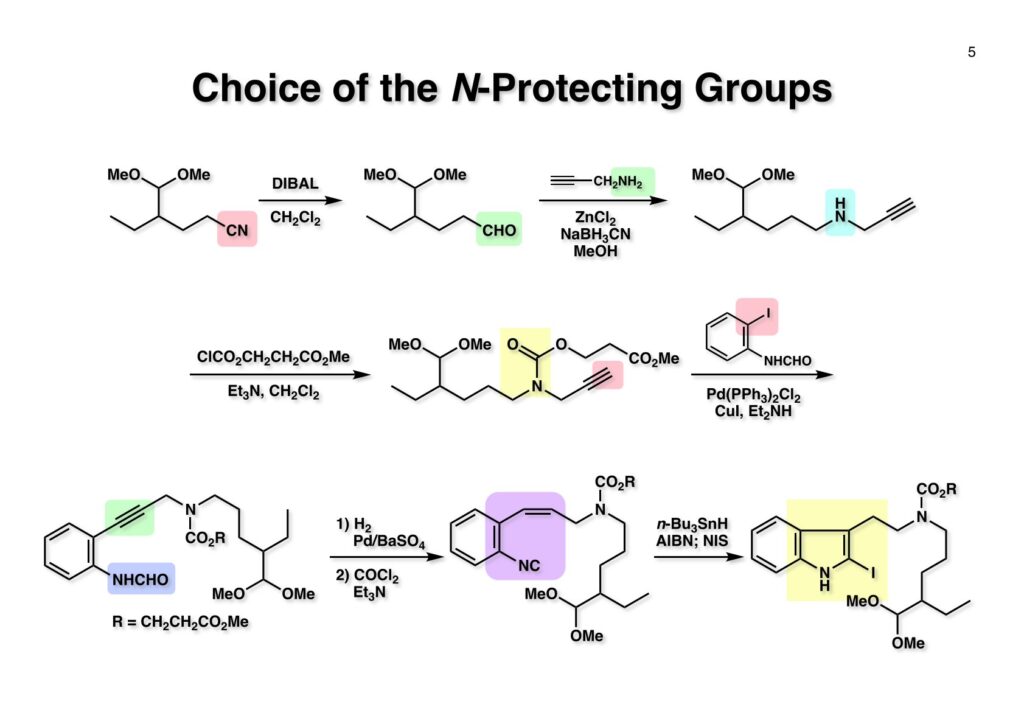
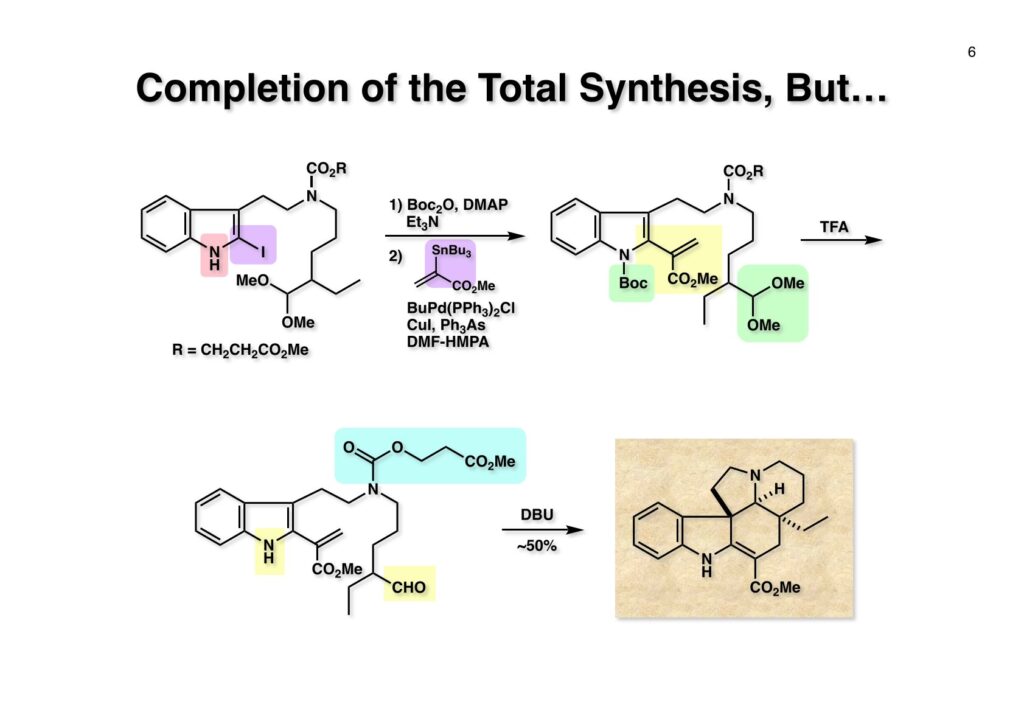
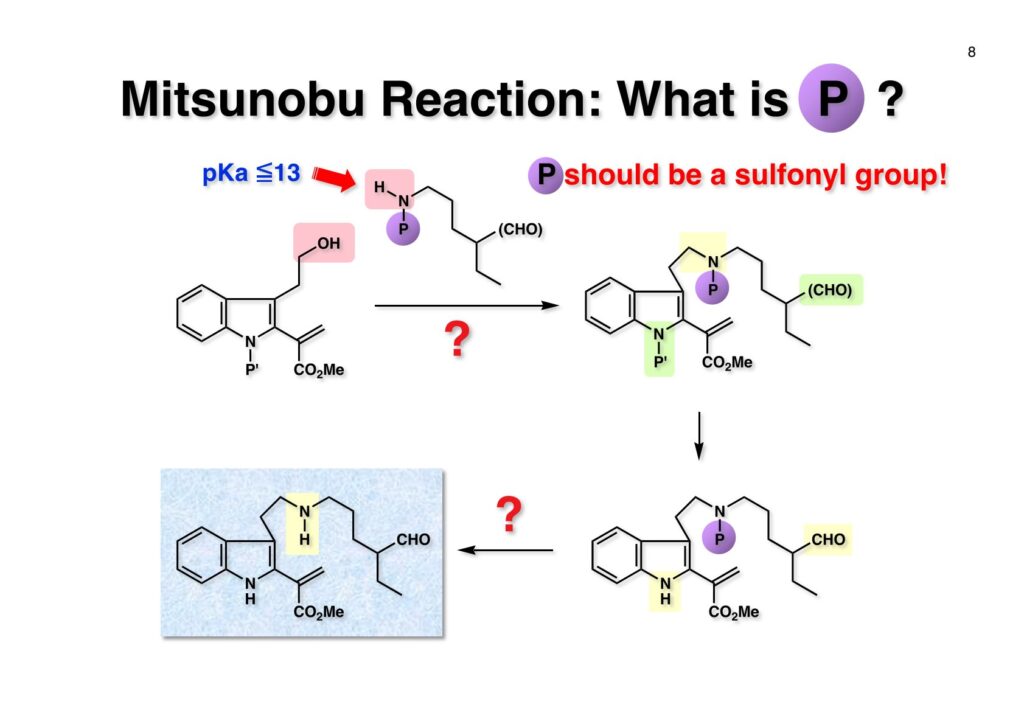
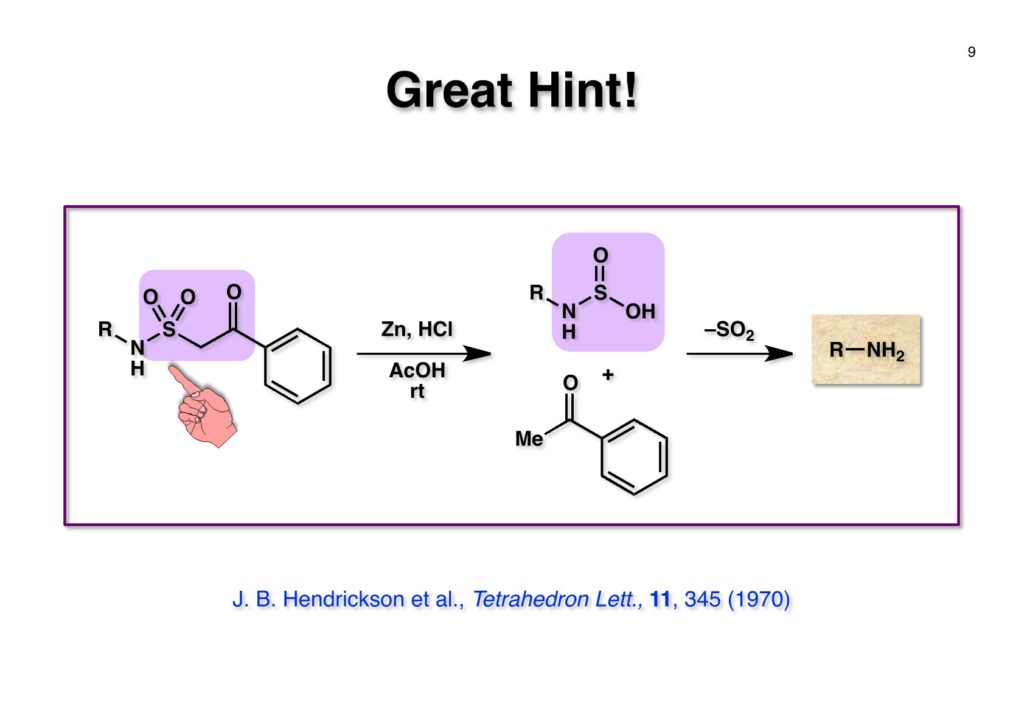
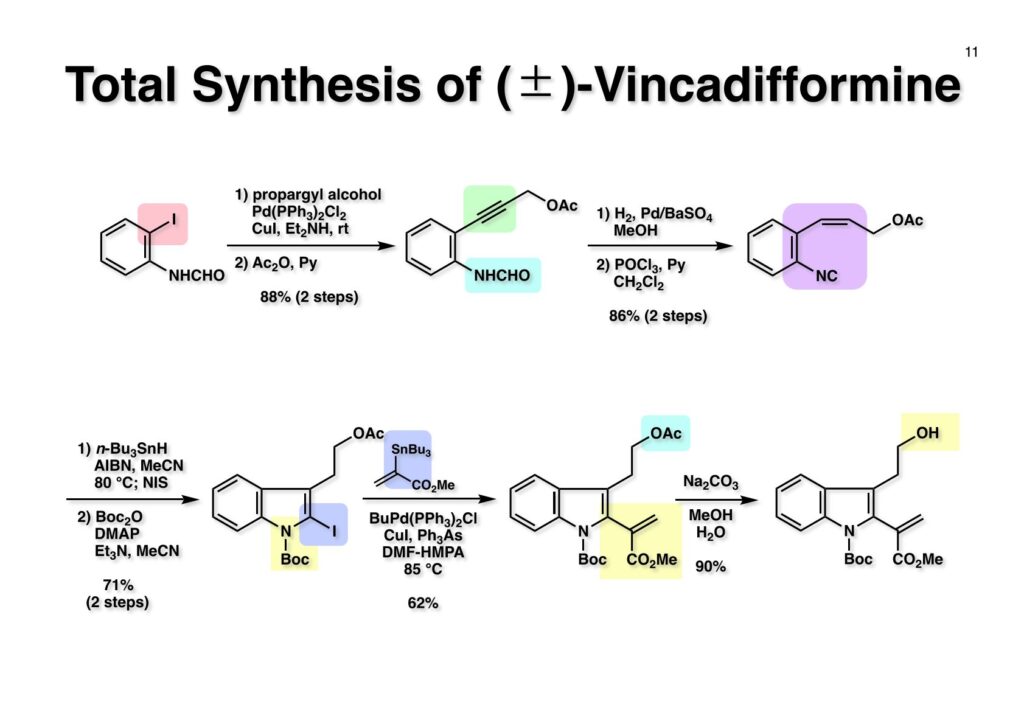
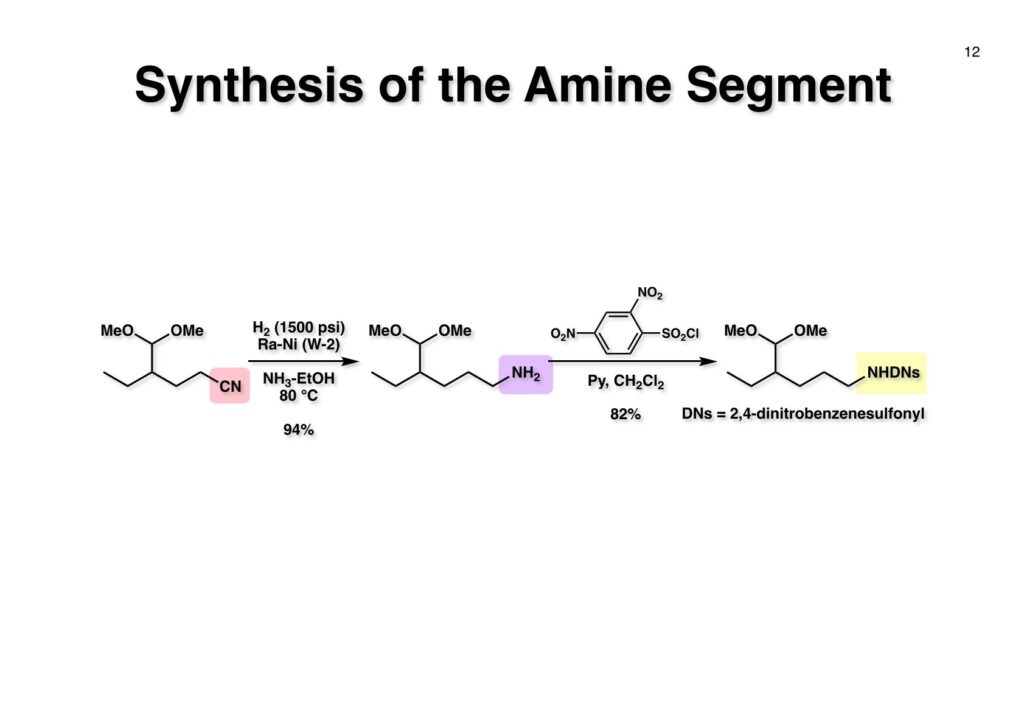
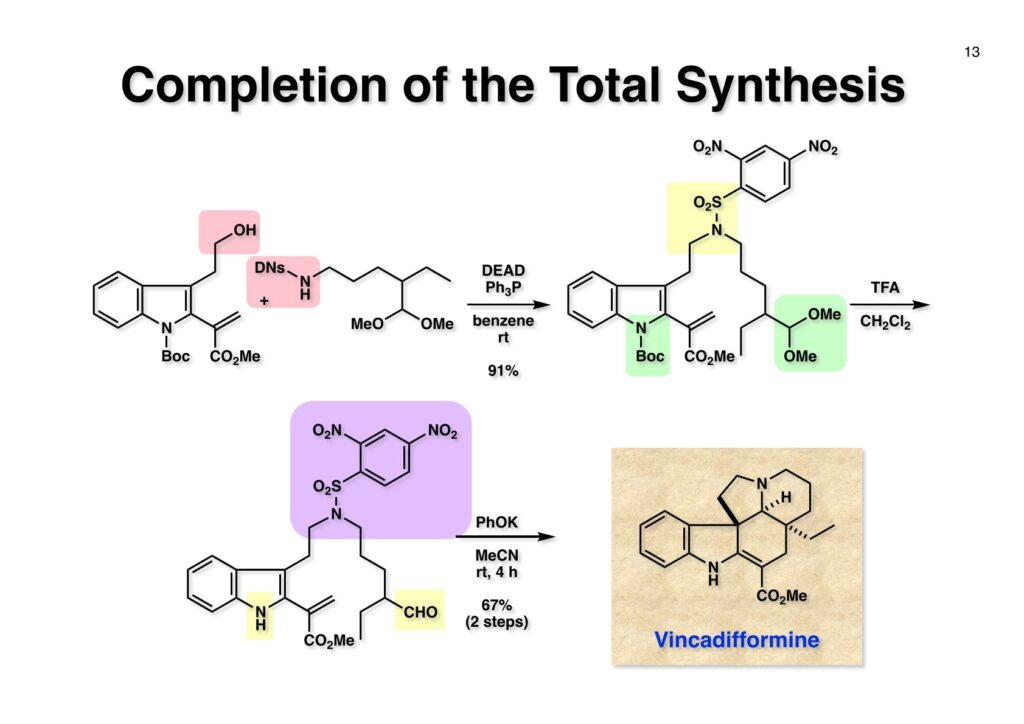
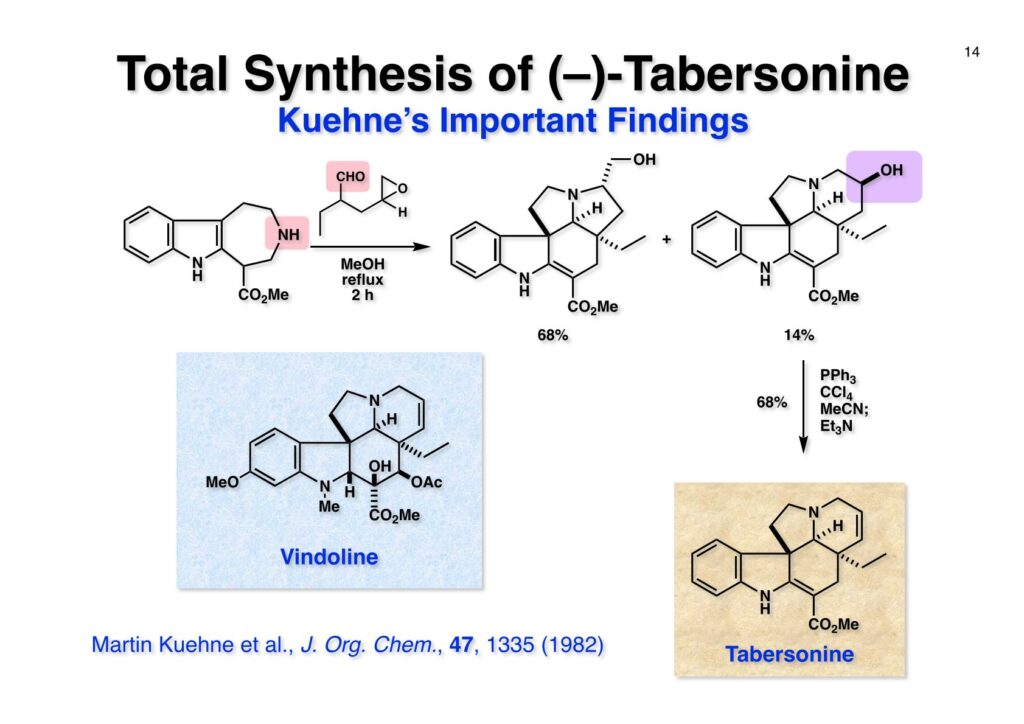
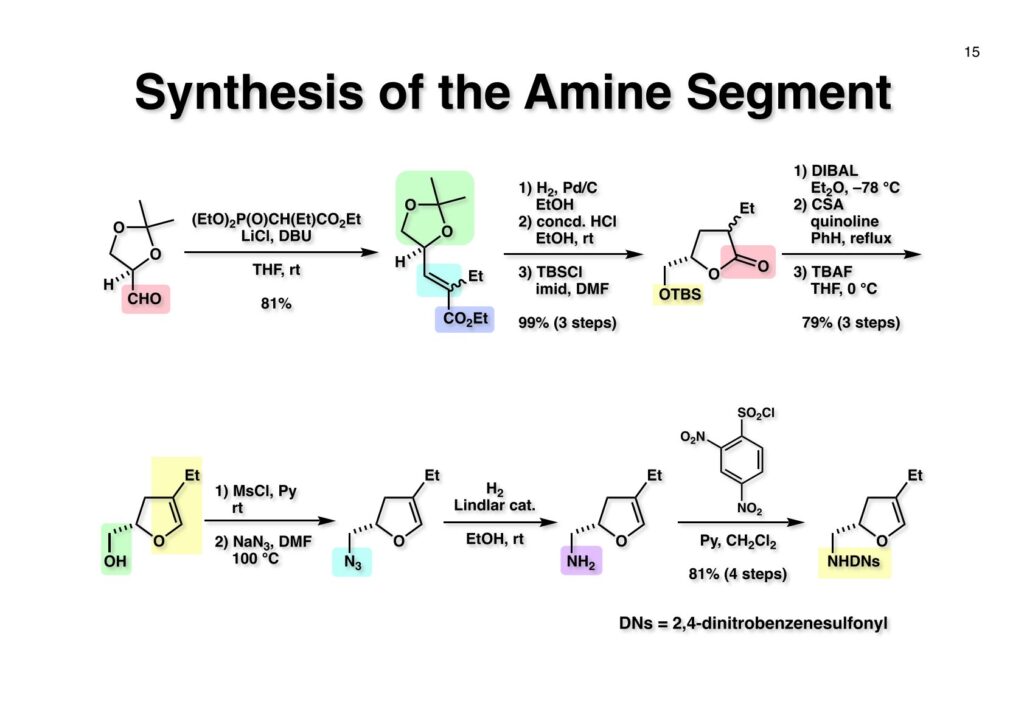
1993年のある日、私はオフィスで院生のXiaoqi Chenと何かは記憶に無いが話をしていた。彼の話にはいい加減に生返事をしていたが、頭の中でモヤモヤとある反応を思いついた。そこですぐに黒板に向かいイソニトリルとオレフィンがオルト位に配置された化合物 (1-1) にラジカル反応を行うとどうなるのかチョークを走らせた。イソニトリルへのラジカル反応は1968年のJACSに当時京大の三枝研の助手だった伊藤嘉彦先生が報告されていた。Cyclohexyl isocyanideにBu3Sn·が付加するとC-N結合が開裂し、結果としてシクロヘキサンとBu3SnCNが生成するというものだ。後にD. H. R. Bartonがアミンをホルムアミド経由でイソニトリルにしてラジカル反応で脱アミノ化をし、Bartonのdeaminationということになってしまった。伊藤先生に「やられちゃいましたね」ってからかったら「当時はそこまで思いが至らなかった」と少し悔しがっておられた。それはともかく、シクロヘキシルラジカルよりはフェニルラジカルの方がエネルギーは高いのでC-N結合の切断は遅いはずだからイミドイルラジカル (1-2) は近傍のオレフィンを攻撃し、速度論的に有利な5員環 (1-3) を形成するはずである。Bu3SnHから水素を引き抜いて生ずる (2-3) は速やかにインドールに芳香環化して2位にスズの入った (2-2) が生成するはずである。これはなかなか面白い反応になると思い、hapalindole Gの全合成を終えたばかりのXiaoqiにちょいと実験をやってくれと頼んだ。最初の実験でインドールが出来るのが分かったが、(2-2) はTLC精製の際にprotodestannylationが起こってスズが抜けてしまった。そこで (2-2) を単離せぬままStilleカップリングを行ったところ容易に2,3-2置換インドール (3-1) が得られた。しかしラジカル反応をStilleカップリングのためにやるのも手間なので (2-2) をヨウ素かNISを使って単離できる (2-1) に変換した。 2-Iodoindole (2-1) は酸性条件では脱ヨウ素化が起きて溶液がヨウ素の色になるが、中性やアルカリ性では安定である。薗頭カップリングで2位にアセチレンが導入できるし (1-1)、一酸化炭素存在下でStilleカップリングを行うとα,β-不飽和ケトン (2-2) になる。この時点でインドールアルカロイドの合成に使えると思い、2-stannylacrylate (3-1) を用いてStilleカップリングを行ったところ目的物 (3-2) が得られた。この反応を2-stannylindoleとmethyl 2-bromoacrylateを使ってStilleカップリングを行ったが非常に低収率であった。これは高温でmethyl 2-bromoacrylateがどんどん分解していったことと、(3-2) が二量化するためである。 Vincadifformine (1-1) はaspidosperma属アルカロイドの中でも比較的簡単な構造を有し、Ernest WenkertやIan Scottによって生合成的にはsecodine (1-2) を経由することが示され、Vermont大学のMartin Kuehneは合成的にもsecodine経由で生成することを証明した。(1-2) のエナミンは (1-3) のアルデヒド-アミンから合成できるので、アミンを保護した (2-3) からStilleカップリングを行えばよい。(2-3) は我々の第一世代インドール合成法を利用するとなれば (2-2) が中間体となる。これを更に遡れば (2-1) に示したような薗頭カップリングということになる。 (1-1) はブチルアルデヒドからStorkのエナミン合成法でアクリロニトリルと反応させ、アルデヒドをジメチルアセタールにして得られた。これをDIBALで還元してアルデヒド (1-2) に変換し、更にプロパルギルアミン (1-3) と還元的アミノ化をして (1-4) を得た。アミンの保護基はBocを一番最初に使ったのだが、TFAで脱保護してエナミンを作る段階で副反応が起きてvincadifformineの収率が非常に低くモノにはならなかった。そこでより穏和な条件で脱保護できる2-carbomethoxyethyl carbamate (2-1) に変換した。次に2-iodoformanilide (2-2) との薗頭カップリングを行って (3-1) を得た。アセチレン (3-1) の部分還元はPd/BaSO4を用いて注意深く水添し、ホルムアミドをホスゲン-Et3Nで脱水してイソニトリル (3-2) が得られた。第一世代のインドール合成法は問題なく (3-2) に適用できて2位にBu3SN基が入ったインドールが出来たが、NISを使ってより安定な2-iodoindole (3-3) に変換した。 (1-1) の2-stannylacrylateとのStilleカップリングはインドールの窒素を保護しないと二量化による収率低下を招くので、まずBoc2O-DMAPでBoc化してからStilleカップリングを実行して (1-2) を得た。次にN-Boc基とジメチルアセタールをTFAで除去して得られた (2-1) の窒素保護基をDBUによるretro-Michael反応で除くとsecodine経由でラセミ体のvincadifformineが生成した。まあ、これで取り敢えず全合成は達成したのだが、どうもダラダラと間延びしているので、もっとconvergentな合成にしてから論文にしたいと思った。 Vincadifformined (1-1) を合成するにはsecodine (1-2) を合成すれば良いことはご承知のとおり。そのためには (2-3) のようなアミン-アルデヒドを構築する必要がある。Convergent合成を目指すとなればインドール部分 (2-1) とアミン部分 (2-2) を結合すれば良いとなる。と、ここまで書いたところ、なぜインドール部分をアルデヒドにしてアミンとの還元的アミノ化を行わなかったのかが思い出せない。おそらくArCH2CHOが不安定で、適時アルコールを酸化して調製しなければならないことを嫌がったのだと思うが…。もし還元的アミノ化ルートを採用していたらノシル基の化学は生まれていなかっただろう。 アミン誘導体とアルコールをカップリングさせる一番マイルドかつ有用な反応は光延反応だろう。確か1971年のハーバード大学化学科のcumulative examination (Cume) でこの反応が出題されていた。図書館でCumeの過去問をチェックした時にDEAD (diethyl azodicarboxylate) が酸化剤となり得る事が全く理解できなかった私には印象的な反応である。この反応の求核剤となるHXのpKaは~13>であると言われていたのでアミドとかカルバメートのpKaは17付近なので不適格である。スルホンアミドのpKaは10付近なので光延反応は可能である。ただ (1-1) と (1-2) の光延反応で得られる (1-3) の保護基を除去した (2-2) でアルデヒドやアクリレートを破壊させずに (2-1) を与えるようなスルホニル基が存在するだろうか?トシル基が無理なのは最初から分かっていたがN-S結合が簡単に切れるようなスルホンアミドは全く頭に浮かんでこなかった。 とにかくアイデアが浮かばなかったので院生がよく使っていたGreene & Wutsの「Protective Groups in Organic Synthesis」でアミンの保護基のスルホンアミドの項に目を通してみた。そこに記載されていたHendrickson (Brandeis大学の教授でWoodward研出身)の研究で頭をガーンと打たれた気がした。(1-1) のフェナシルスルホンアミドの除去に亜鉛-塩酸を使っていたのだ。それまでN-S結合を切ることしか頭に無かったのに、ここではC-S結合を切っていたのだ。その頃Ullmannカップリングの本を読んでいた私の頭にはニトロ基が充満していたのですぐにMeisenheimer Complexが思い浮かんだ。そこで次ページのようなアミンの活性基を思いつき、これはName Reactionになる予感がしたのは事実である。因みにこの本の著者のTheodora GreeneはMITの教授だったFrederick Greeneの奥さんで、子供を育てながらCorey研の大学院生をやっていた。セミナーなどで、ちょっと院生にしては年がいっているなー、とは思っていたけど。Peter Wutsはミシガン大学の助教授だった頃、ロッキー山脈のPingree Parkで開催されたNSF Workshopでライス大学の助教授だった私と同室になり仲良くなった間柄だ。その後彼はUpjohnのプロセスケミストとなり、私もミシガンのUpjohnにコンサルティングで年2回訪問していたので旧交を温めた。今でも年末にはメールで無事を確かめ合っている仲である。 前ページの閃きで最初に思いついたのは2,4-dinitrobenzenesulfonamide (2-1) でこのNHはpKaが8くらいだろうから光延反応やK2CO3を塩基としてアルキルハライドでアルキル化できるだろう。(1-1) に着目してみると、ベンゼン環に強力な電子吸引基である二つのニトロ基と一つのスルホニル基が存在している。このような電子欠乏なベンゼン環にはsoft nucleophileが付加してMeisenheimer complex (1-2) を生成するということが今は知らないが昔の有機化学の教科書には書いてあった。付加する場所としてエネルギー的に有利なのは1位、3位、5位だろうが、3位と5位に付加しても求核種が退去するしか道は無い。ところが1位にたとえばチオレートが付加した場合、NSO2-の方がRS-よりも安定なアニオンで脱離能が大きい。生じた (1-3) はプロトン付加後にSO2を放出して2級アミン (1-4) が得られるという仕組みだ。この反応は最初の実験で成功し、その後大きな広がりを見せた。2,4-Dinitrobenzenesulfonamideは非常に反応性が高く、アミンやNaBH4でも脱離してしまうので、活性化基を長持ちさせるためにはニトロ基を一つ減らしたo- or p-nitrobenzenesulfonamideが一般的である。ノシル基の化学は全合成の項を書き終わってから反応開発の項で詳述するつもりである。 o -Iodoformanilide (1-1) とプロパルギルアルコールとの薗頭カップリングの後にアルコールをアセチル化して (1-2) を得た。アセチレンの部分還元はPd/BaSO4を触媒にして過還元が起きないようにcis -オレフィンに変換し、ホルムアミドを塩化ホスホリルを用いて脱水しイソニトリル (1-3) を得た。この脱水反応はホスゲンを用いた方が短時間かつ高収率になるが、ホスゲンを実験室で用いるのは特に日本では容易では無いのでPOCl3を用いた方が良い。他にCCl4-PPh3-Et3Nという条件もあることは知っておいて損はない。(1-3) をn -Bu3SnH-AIBNでラジカル反応を行い、反応液にNISを加えて単離可能な2-iodoindoleに変換した。次にBoc2O ~DMAPでインドールのNHをBoc化して (2-1) を得た。Boc化しておかないと光延反応の条件でシクロプロパン化が起きてしまい分子間反応が進まないからだ。また、次の (2-2) とのStille couplingで (2-3) の二量化が起きるのを防ぐ目的もある。(2-3) のアセテートを加水分解してインドール部分 (1-4) の構築ができた。アミン部分の合成は、まずニトリル (1-1) のRaneyニッケルによる接触還元でアミン (1-2) に変換した。ここで高濃度のアンモニアを混在させるのは昔からの手法で二級アミンの生成を抑えるためだ。つまりニトリルの還元の中間体であるイミンが一級アミンと縮合して、さらに還元が進行すると二級アミンになってしまうからだ。アンモニアを競争的に反応させてそれを防ぐという理屈である。アミン (1-2) を2,4-dinitrobenzenesulfonyl chloride (1-3) と反応させてアミン部分 (1-4) が用意できた。 インドール部分 (1-1) とアミン部分 (1-2) は光延反応を室温で行うことで収率良く (1-3) を与えた。光延反応は室温で進行することが殆どで、加熱するといけないと思っている人も居るかもしれないが、なかなか進行しないケースで90 °Cまで加熱した事があるので心配することはない。(1-3) のBoc基とアセタールをTFAで除去してアルデヒド (2-1) が得られた。ここではチオールを使ってDNs基を外すことはできない。何故ならばアクリレートへのMichael付加が瞬時に起きてしまいα,β-不飽和エステルでなくなってしまうからだ。そこでMichael付加が起こりにくい酸素系求核種であるPhOK (結晶) を使うことにした。(今ではピロリジンで室温5分でDNs基が外れることが分かっている)アセトニトリル中15当量のPhOKを加えて4時間室温で撹拌することで (1-3) から67%の収率でラセミ体のvincadifformine (2-2) が得られて全合成が完了した。 三共株式会社から小林聡さん(東北大薬出身)が研究生第1号としてグループに加わったので第一世代インドール合成法を使ってvindolineの全合成をやってもらうことにした。光学活性体を作りたかったので以前JOCに報告されていたMartin Kuehneの反応を参考にした。彼の場合はラセミ体ではあるが、アミン (1-1) とアルデヒド (1-2) をメタノール中で加熱環流すると (1-3) と (1-4) が各々68%と14%の収率で得られるということだった。中間体のアミンによるエポキサイドの開環が6員環より5員環形成が有利だったのだが、ここで重要な知見は (1-4) の二級水酸基の立体化学が5環性化合物の立体化学を全て制御している点である。(1-4) の脱水反応でtabersonineが出来ることをKuehneは報告している。光学活性vindolineを合成するモデル実験としてまず光学活性tabersonineの全合成をやってみることにした。 D-Mannitolから2段階で容易に得られるグリセルアルデヒドのアセトナイド (1-1) をRoush-正宗によるHorner-Emmons反応の改良法で (1-2) のジアステレオマー混合物を得た。(1-2) の二重結合を水添し、塩酸/EtOHでアセトナイドを除去すると、より安定な5員環ラクトンが生成し、水酸基をTBS化して (1-3) を高収率で得た。次にラクトンをDIBAL還元してラクトールに変換し、CSA-quinoline存在下ベンゼン中で加熱すると脱水してジヒドロフラン体が生成する。このTBS基をTBAFで除去して (2-1) が得られた。(2-1) は2段階でアジド (2-2) に変換し、二重結合を還元しないようにLindlar触媒を使ってアジドをアミン (2-3) に還元した。アミン (2-3) を2,4-dinitrobenzenesulfonyl chloride (2-4) と反応させてアミン部分 (2-5) が得られた。 (1-1) と (1-2) を光延反応条件に付して95%の収率で (1-3) を得た。これをTFA処理するとBoc基の除去とエノールエーテルの水和でラクトール (2-1) が得られた。今回はピロリジンを使ってDNs基の除去を行い、メタノール-MeCN中で加熱して5環性化合物 (2-2) を単一の生成物として得た。Kuehneの実験結果どおり二級水酸基の立体化学が全てをコントロールした。最後に水酸基の脱水をPPh3-CCl4と加熱することで行い光学活性 (–)-tabersonineの全合成が完成した。