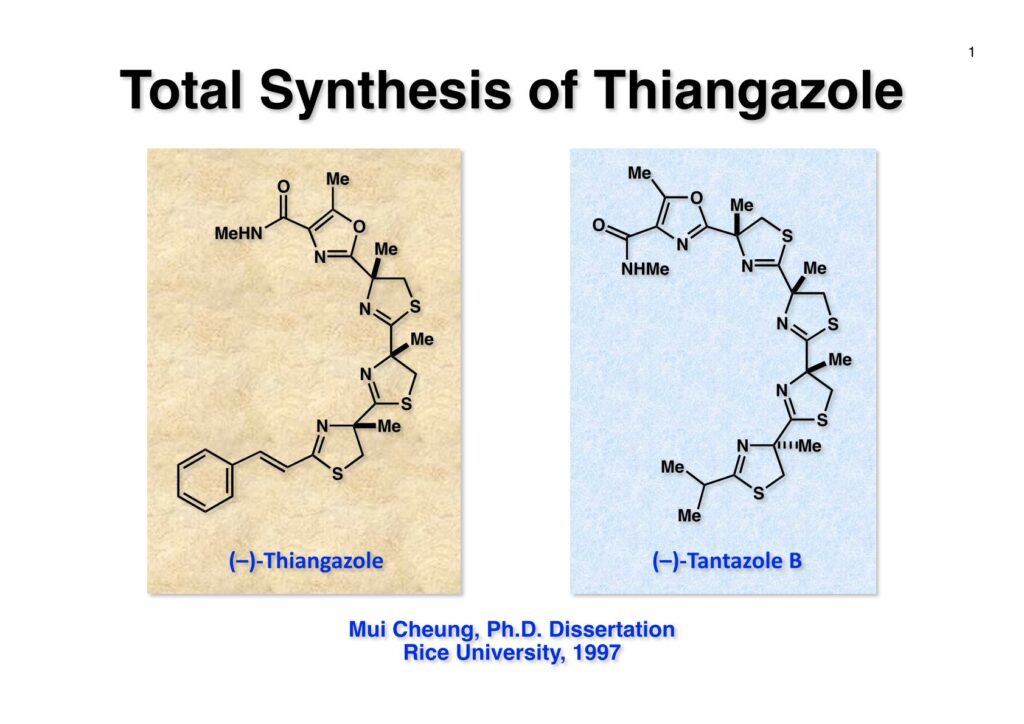
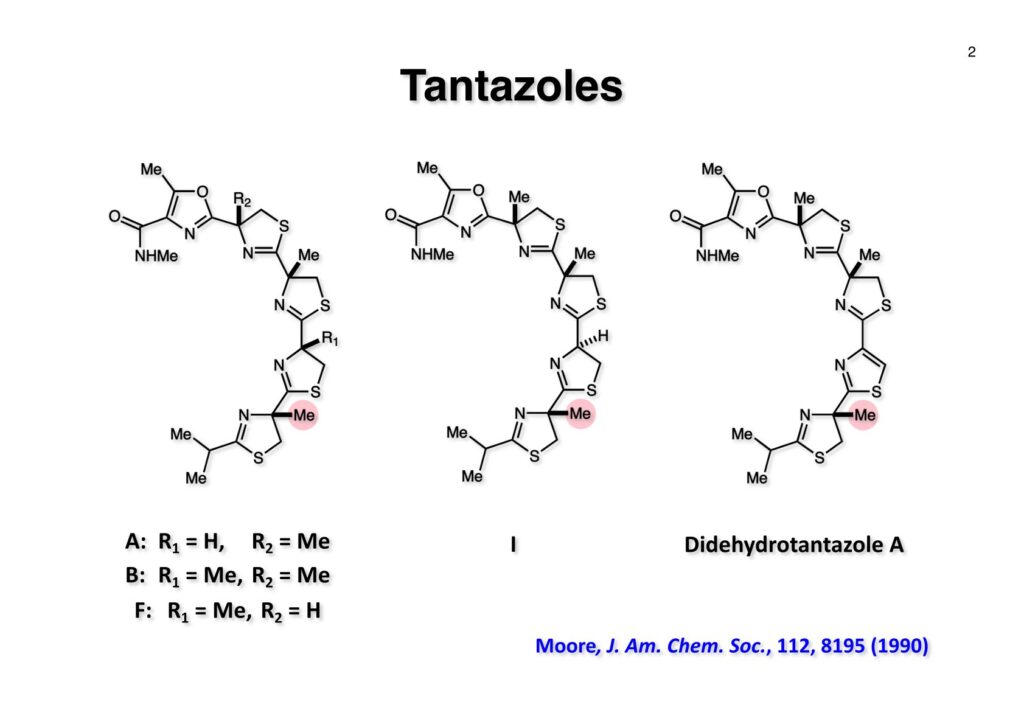
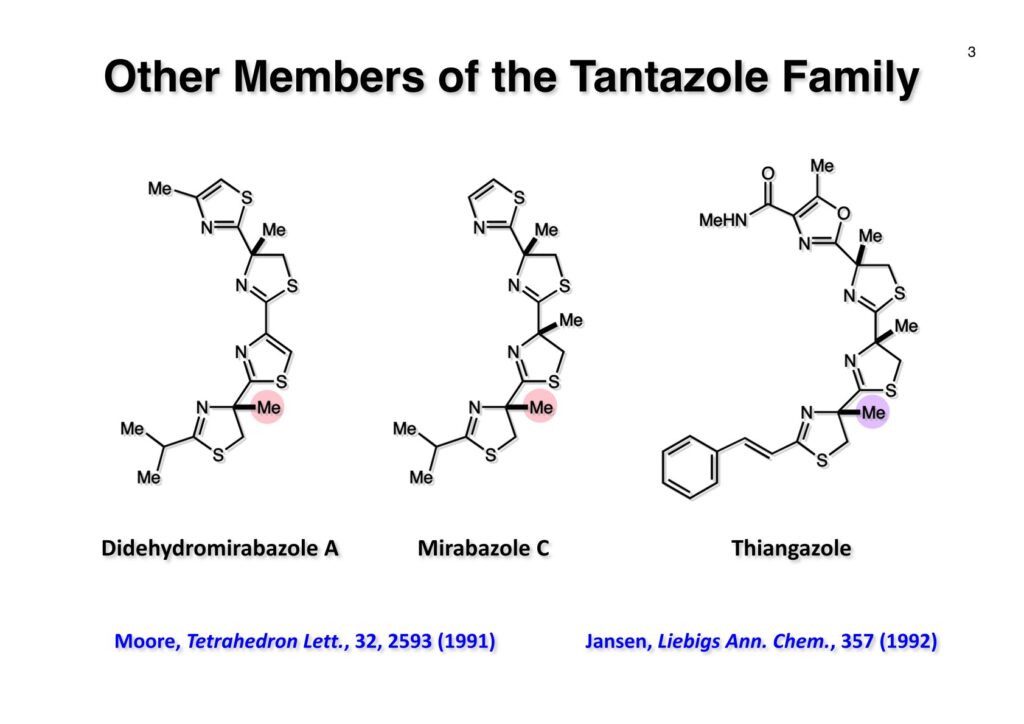
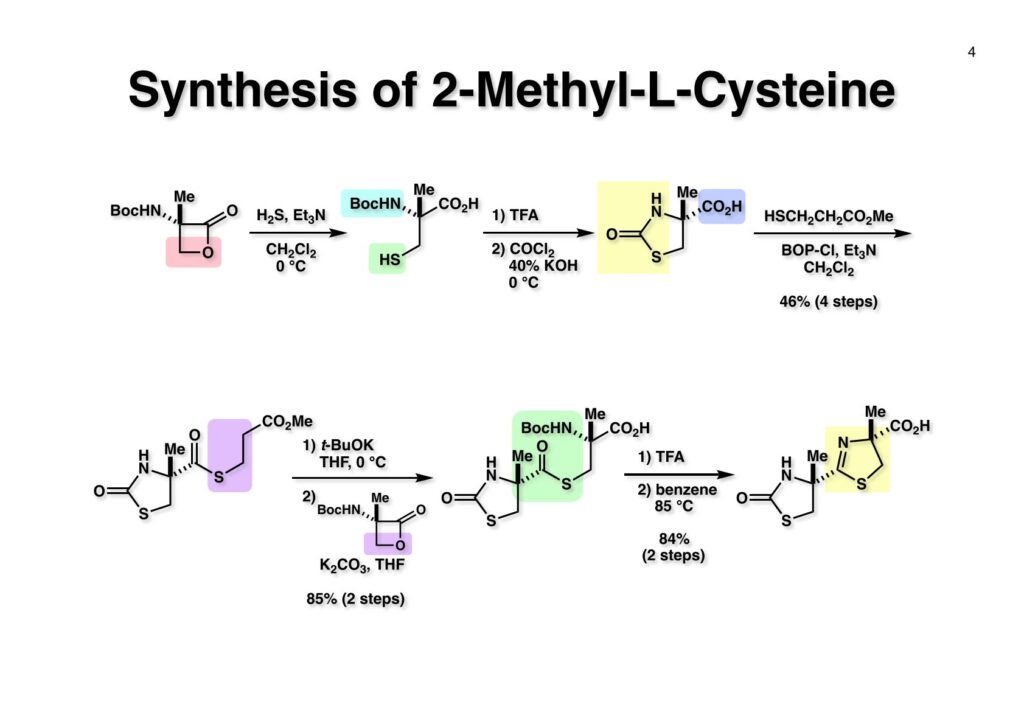
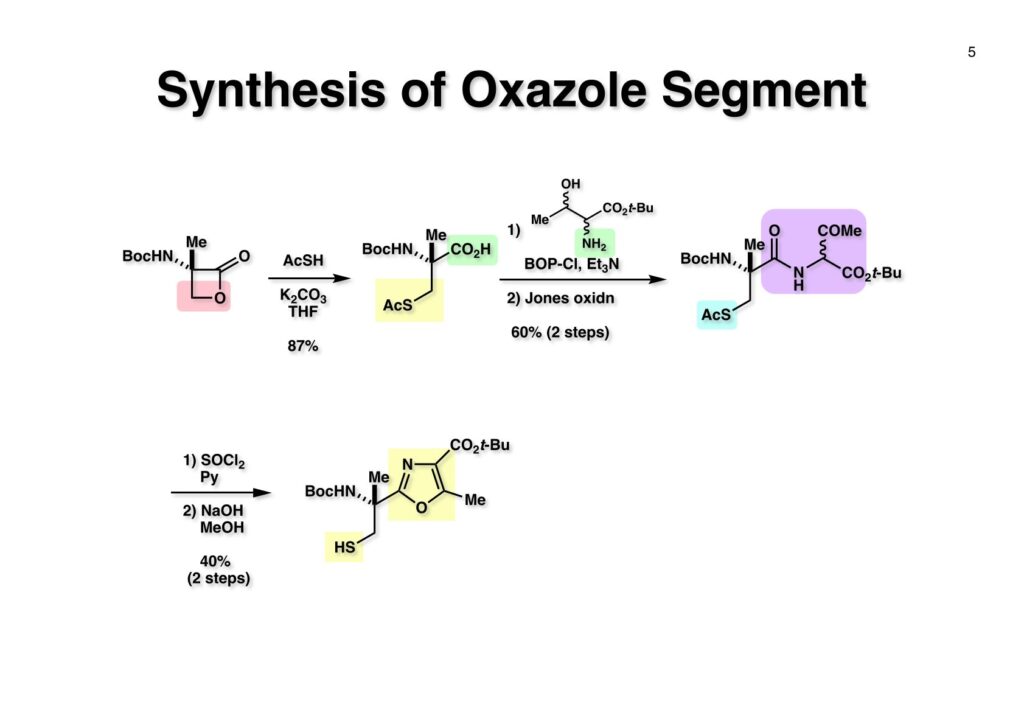
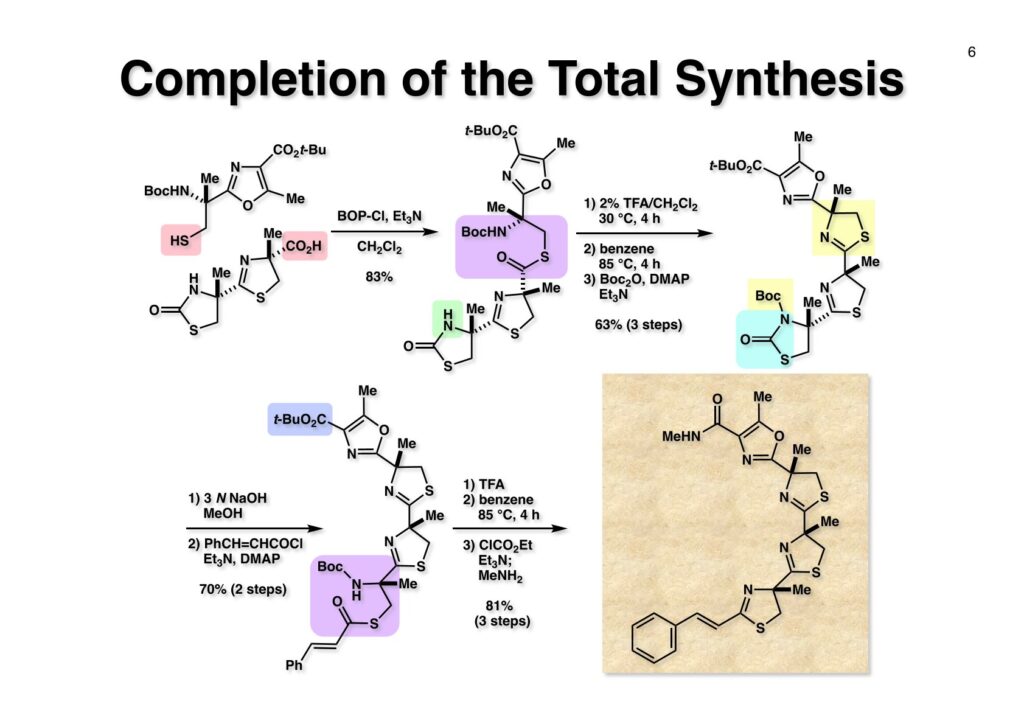
Safracin Bに続いてMui Cheungが全合成を終えたのがthiangazoleである。私が東大に移る前に開始して、移ってからMarco Ciufoliniの指導の下に行ったのだが、例によって日本でガタガタしている時で、後で論文を出そうと思いながら延び延びになって結局日の目を見なかった。2つも論文を出さなかったので、私がMuiに対して終生負い目をおっているのも無理はない。しかし、thiangazoleの合成はtantazole Bの合成を少し変更しただけなので、なかなか論文を書く気になれなかったというのが本音である(説得力のない言い訳ではあるけど)。Thiangazoleはドイツで発酵産物として単離構造決定され、HIVウィルスに抑制効果があると報告された。早速いくつかのグループが全合成を開始したのだが、後にそれほどHIVに対しては効果が無いということが判明し、「なーんだ」という結末になった化合物である。
Mui Cheung, Ph.D. Dissertation, Rice University, 1997.