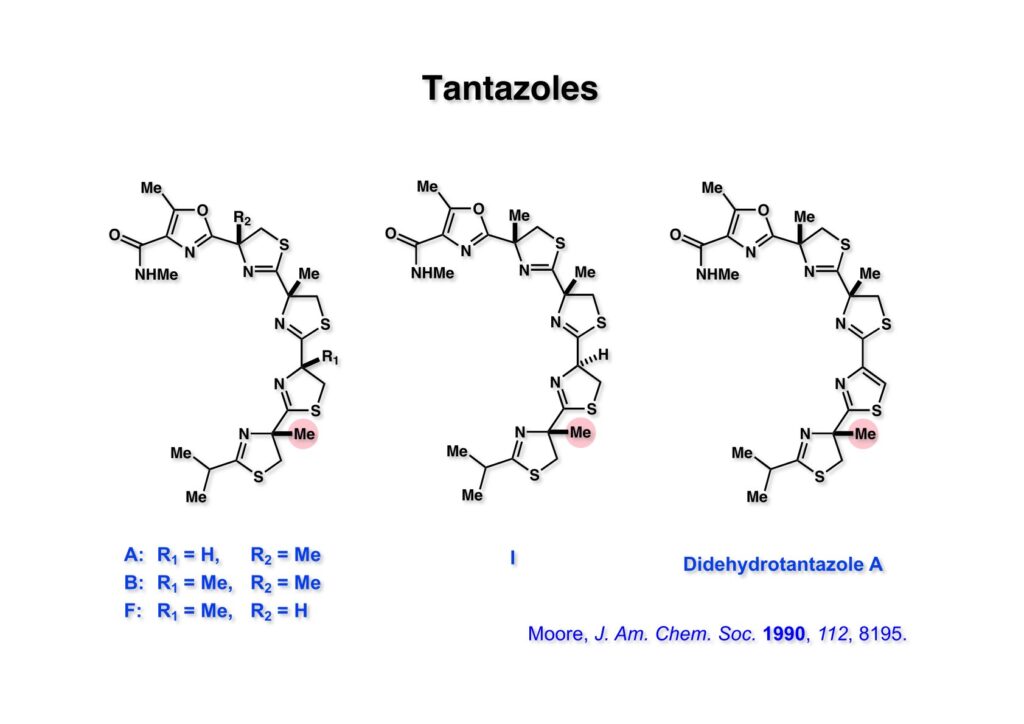
おそらく1989年のGordon Conference on Natural Productsに出席した時だと思うが、ハワイ大学のRichard (Dick) Mooreが新規海洋天然物の講演をした時に、珍しい構造を見て、へーっと思った。彼は下図のように縦型でなく、横型に構造式を書いていたので、なんかネックレスみたいな化合物だという印象を受けた。と言っても、すぐに合成してみようとは思わす。グループミーティングのプロポーザルにtantazole Bを提案して、学生たちがどのように合成しようとするのかを見ていた。もちろん学生のアイデアを盗もうなんてケチな考えからではない。そのうちLianhong Xuという女子学生が FR-900482の全合成を終えたので、もう博士号を取れるから論文を書く準備をしたら、と言ったら、もう少し全合成をやりたいという希望を私に言った。彼女は南開大学出身の優秀な学生で、もう一仕事したいなんて熱心な奴だと思い、tantazole Bの全合成をやってみようか、ということになった。しかし、後から考えると、一学年下のXiaoqi Chen (USTC出身)と恋仲で、もう少し一緒に居たいって、というのが真相のようだ。鈍感?な私は彼らが付き合っていることを全然知らなかった。LianhongはPh.D.取得後にAbbottに就職し、その後、まだタミフルを上市する前のGileadに転職し、その時にストックオプションを貰っているので、私の弟子の中では一番のお金持ちになったと思う。
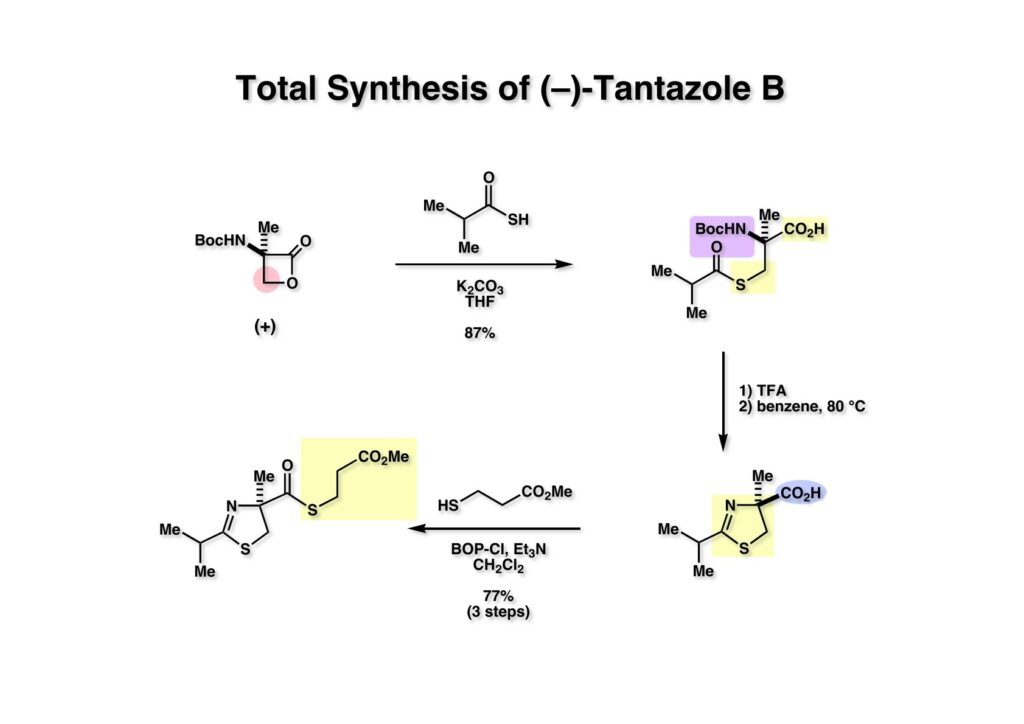
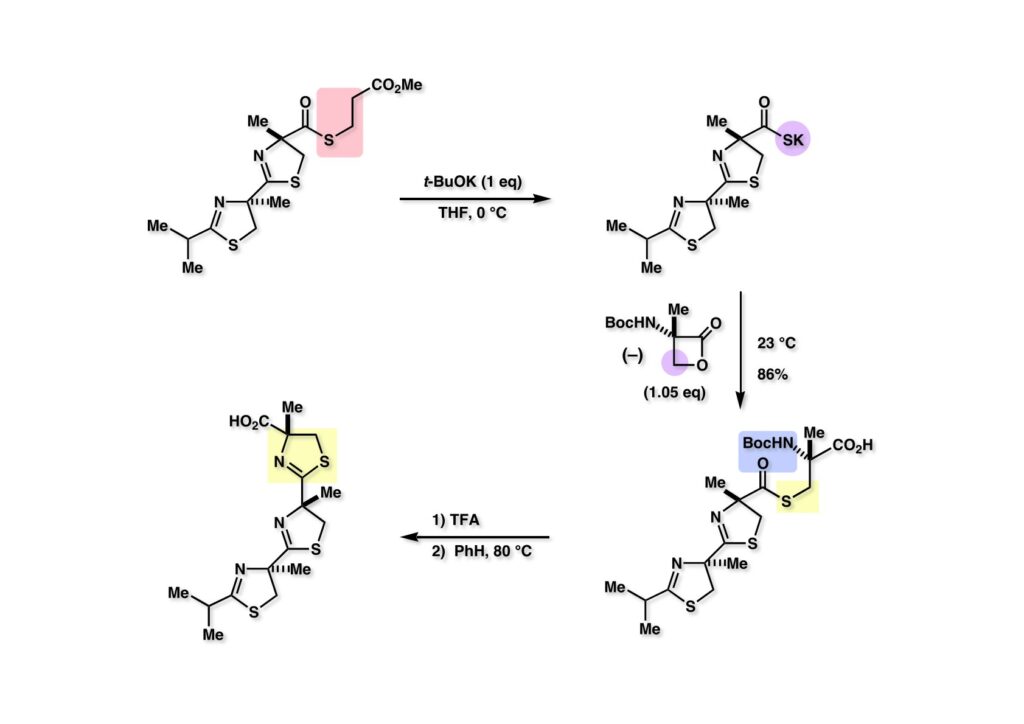
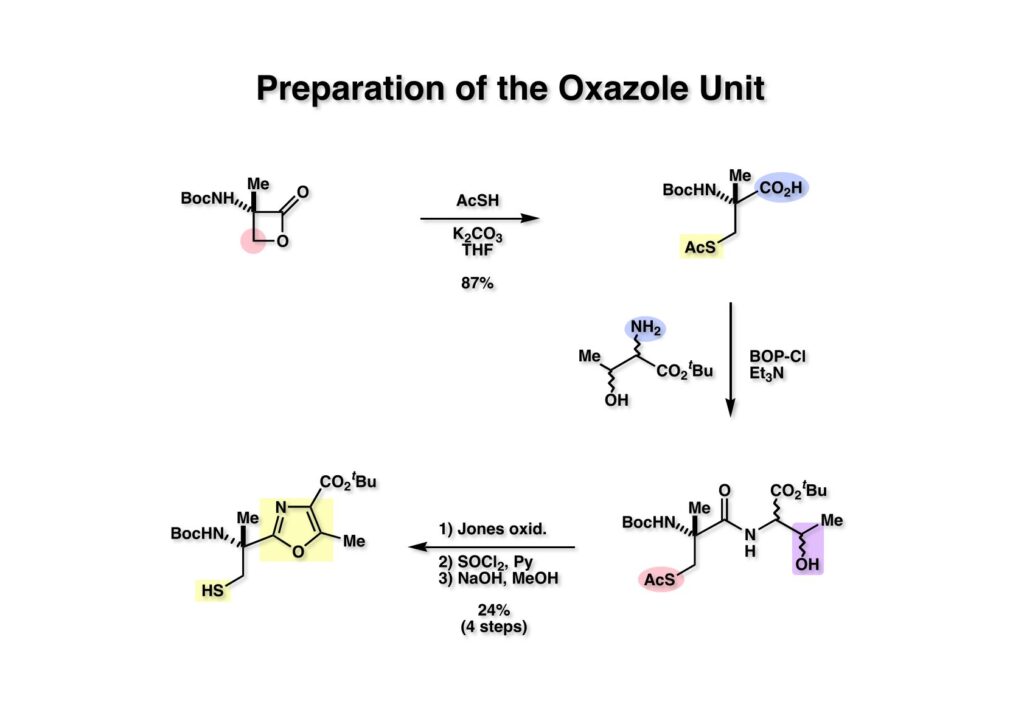
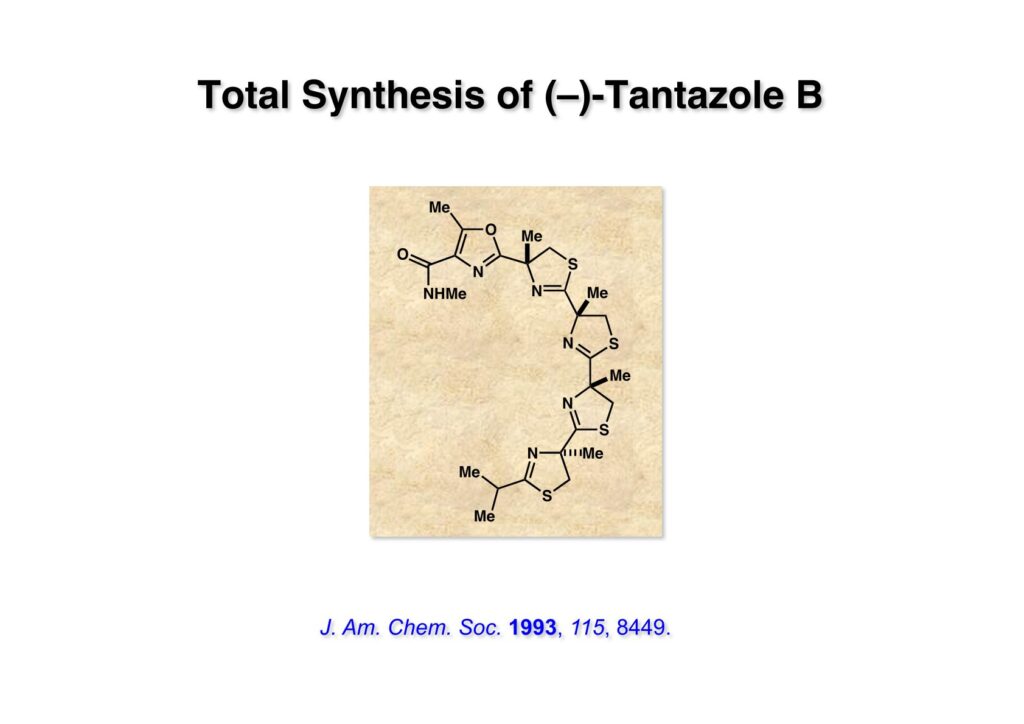
“Total Synthesis of (–)-Tantazole B,” T. Fukuyama and L. Xu, J. Am. Chem. Soc., 115, 8449 (1993).

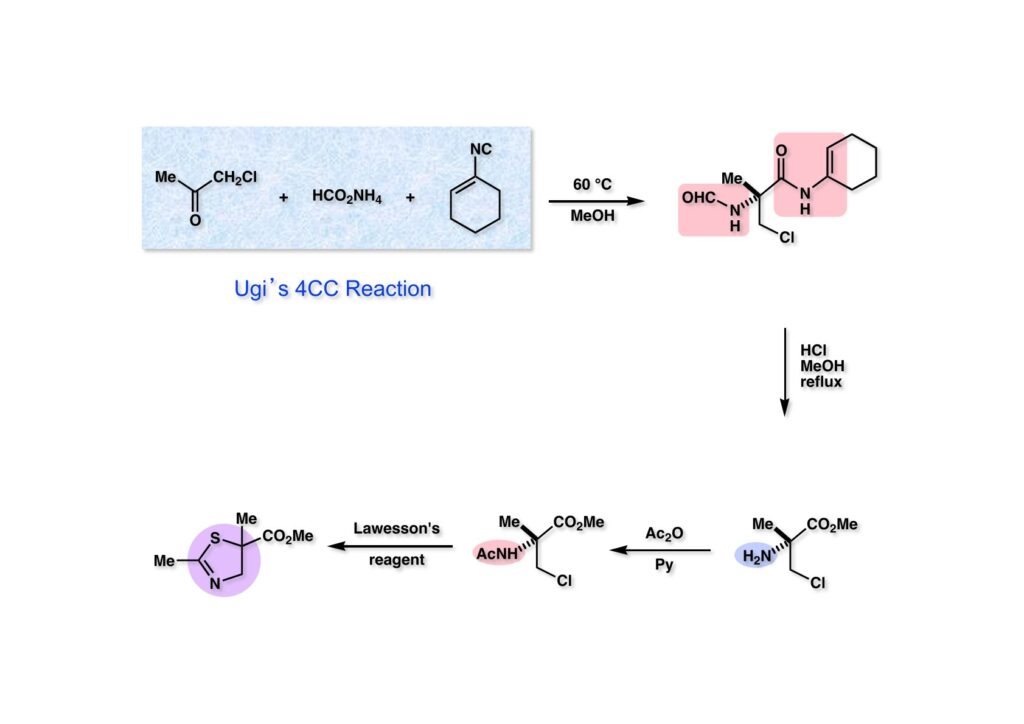
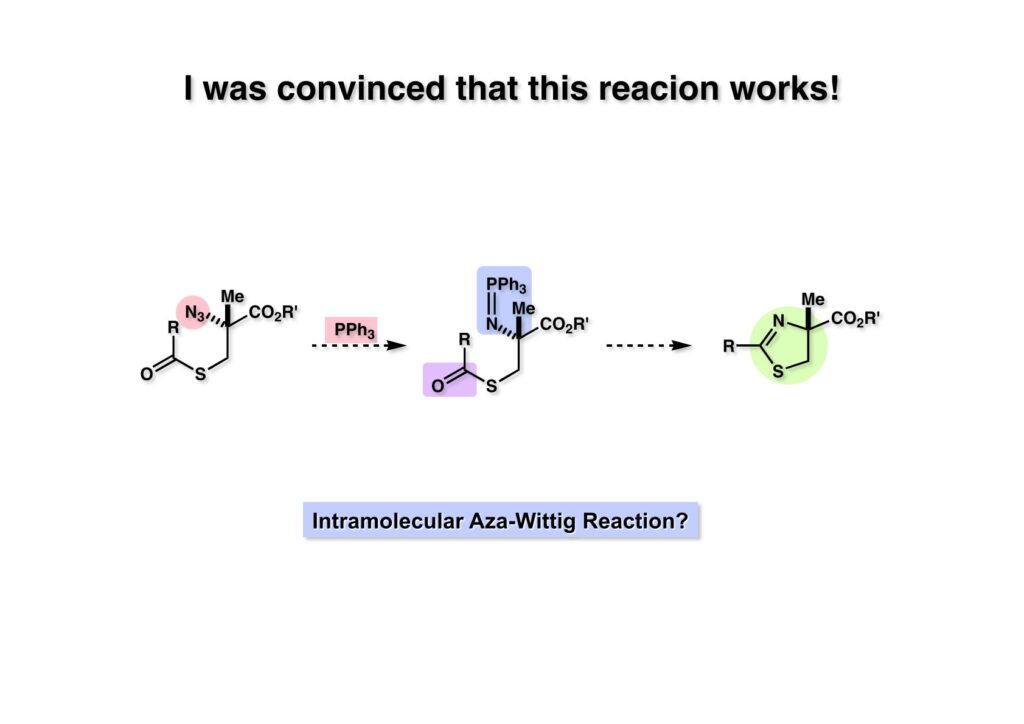

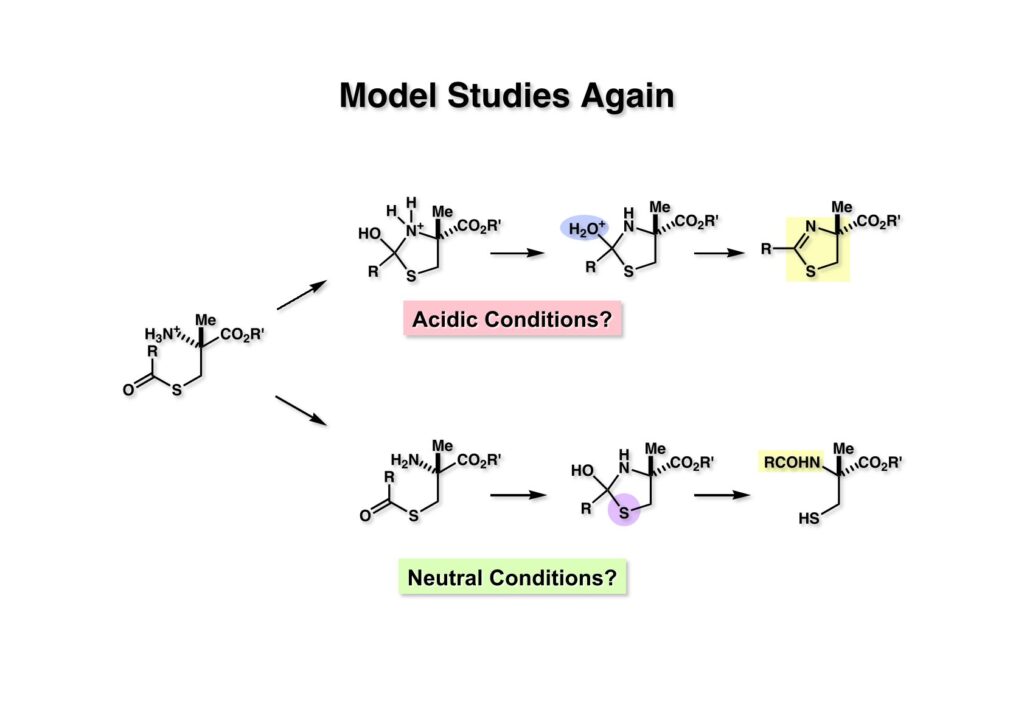
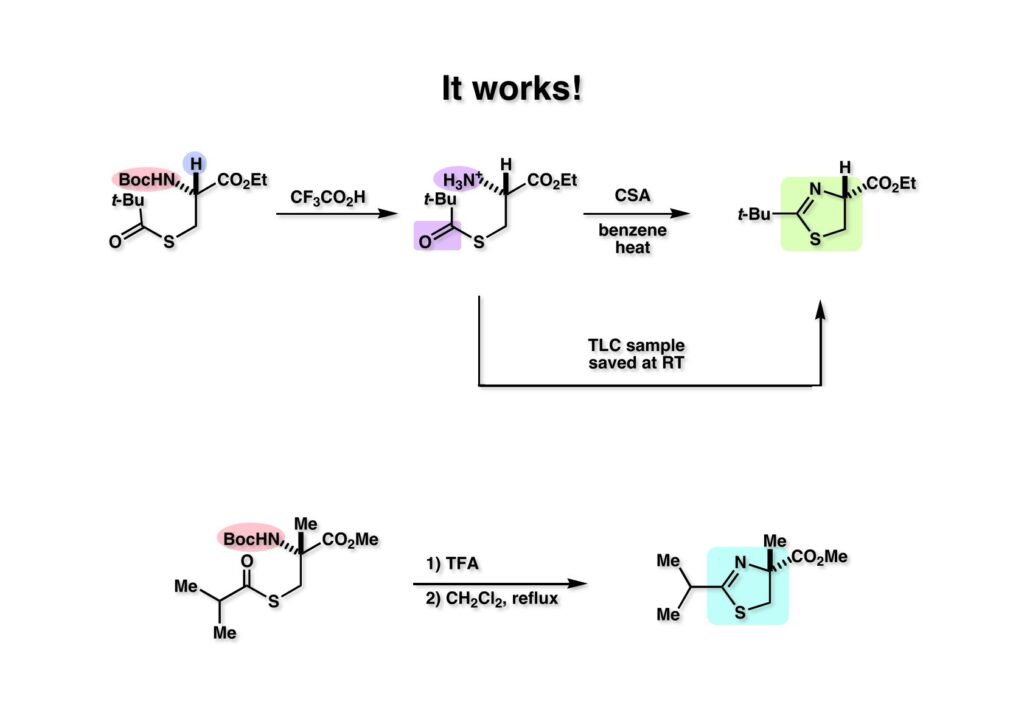
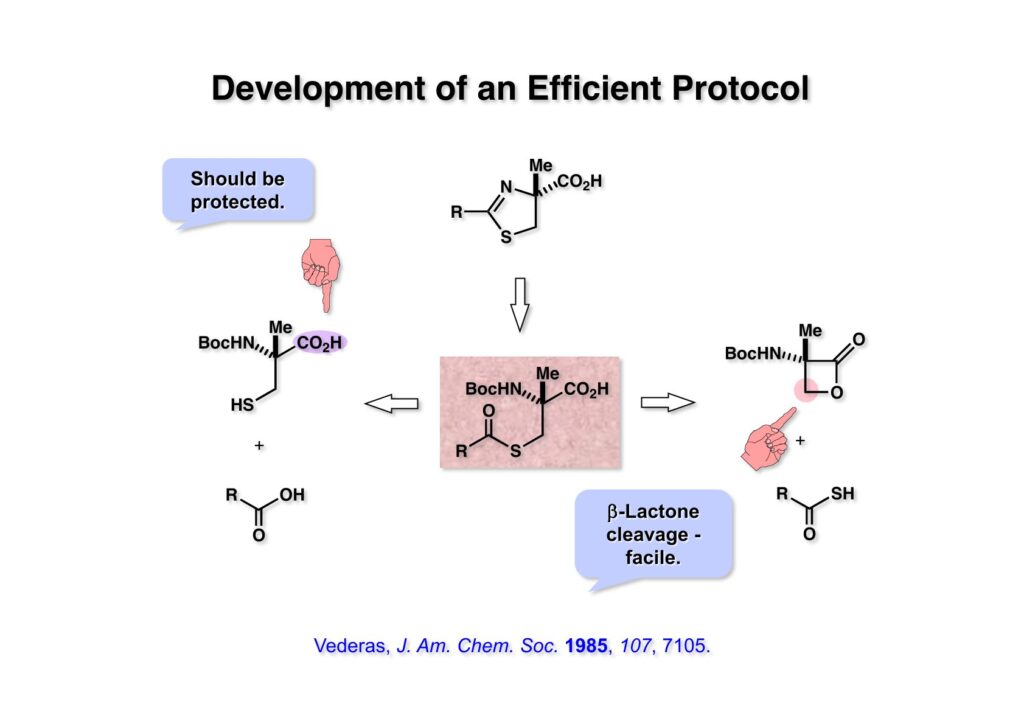
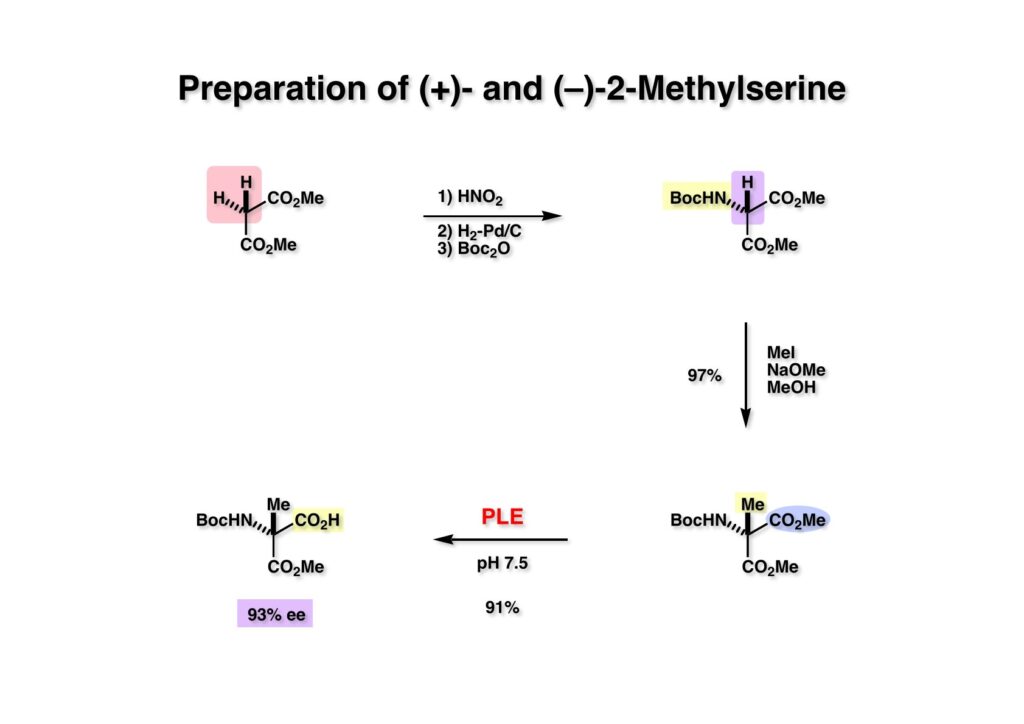
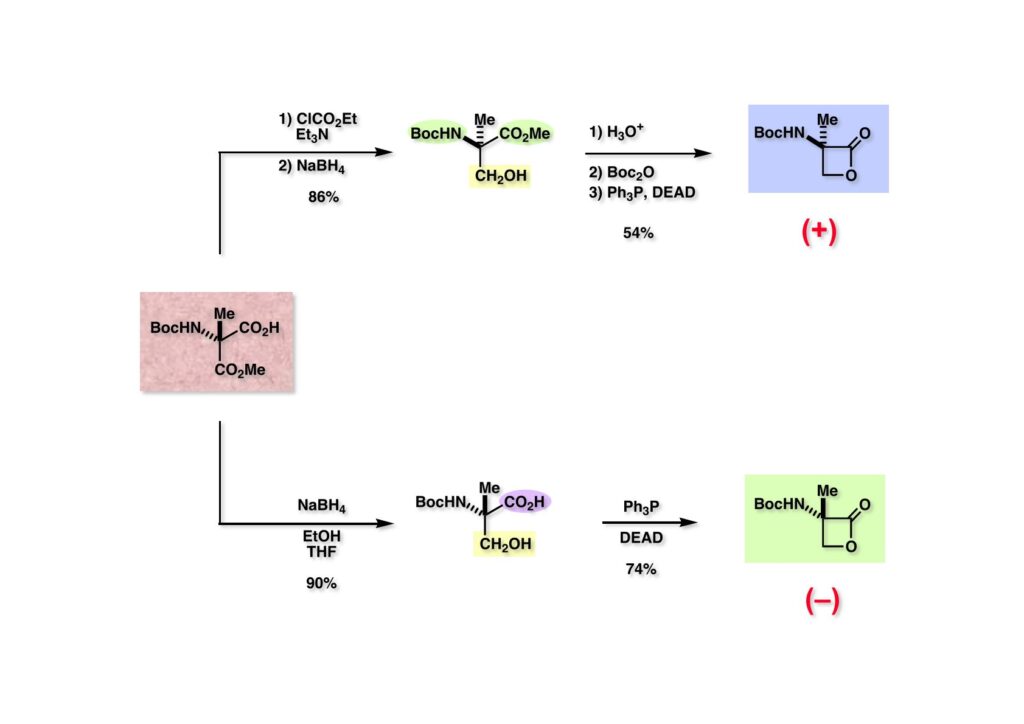
文献既知の方法で (1-1) をNaNO2-HClでニトロソ化(直ちにオキシムに異性化)し、接触還元でアミンに変換したのちにBoc基で保護して (1-2) を得た。(1-2) のCHのpKaはNHに比べて4〜5低いのでNaOMeを用いるとCHだけ脱プロトン化されて容易にメチル化された (2-2) が得られる。ここで酵素の力を借りて光学活性なカルボン酸 (2-1) に変換した。1970年代の有機化学界では酵素を用いると「卑怯者」とされるような風潮があったが、まあ今でも酵素を使わずに済むなら、という雰囲気は残っているかも。ここで使ったPLE (porcine liver esterase) は豚の肝臓をミキサーにかけてアセトンパウダーにしたものだ。もっと精製した酵素になると値段は跳ね上がる。抽出操作で精製したカルボン酸 (2-1) の光学純度は93% eeで、CH2Cl2-hexaneで結晶化して純品が得られる。