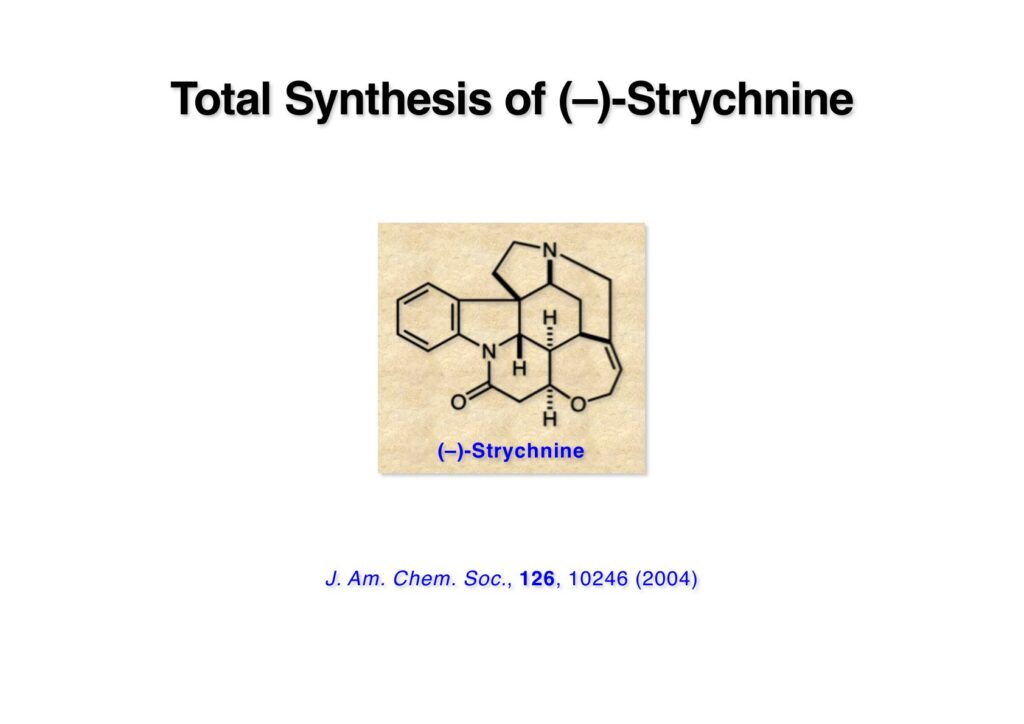
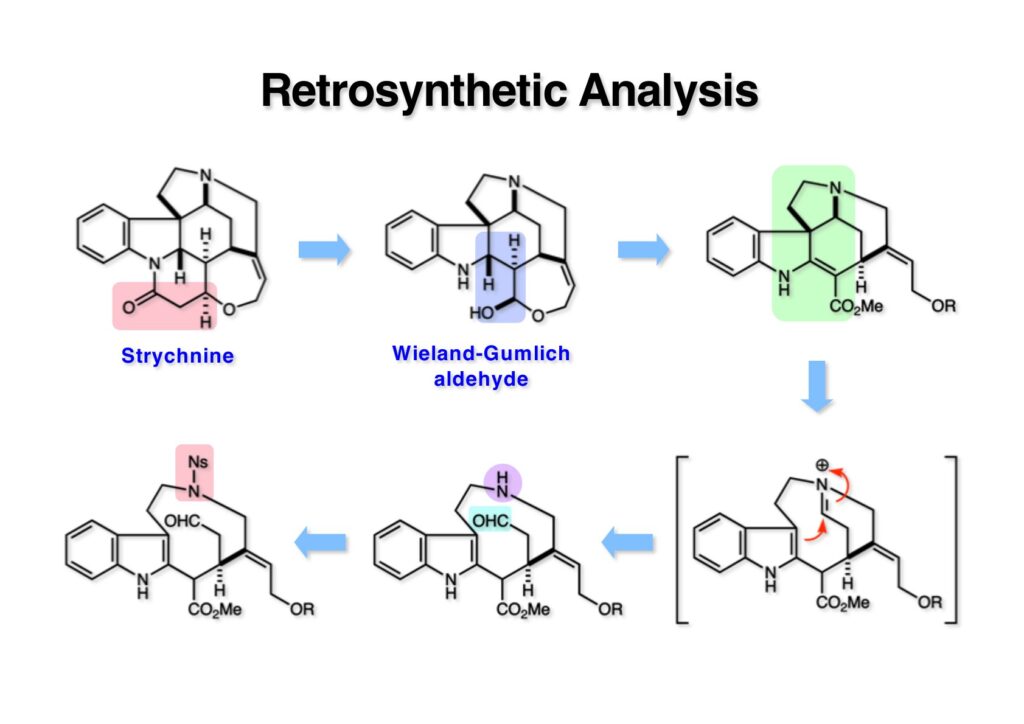
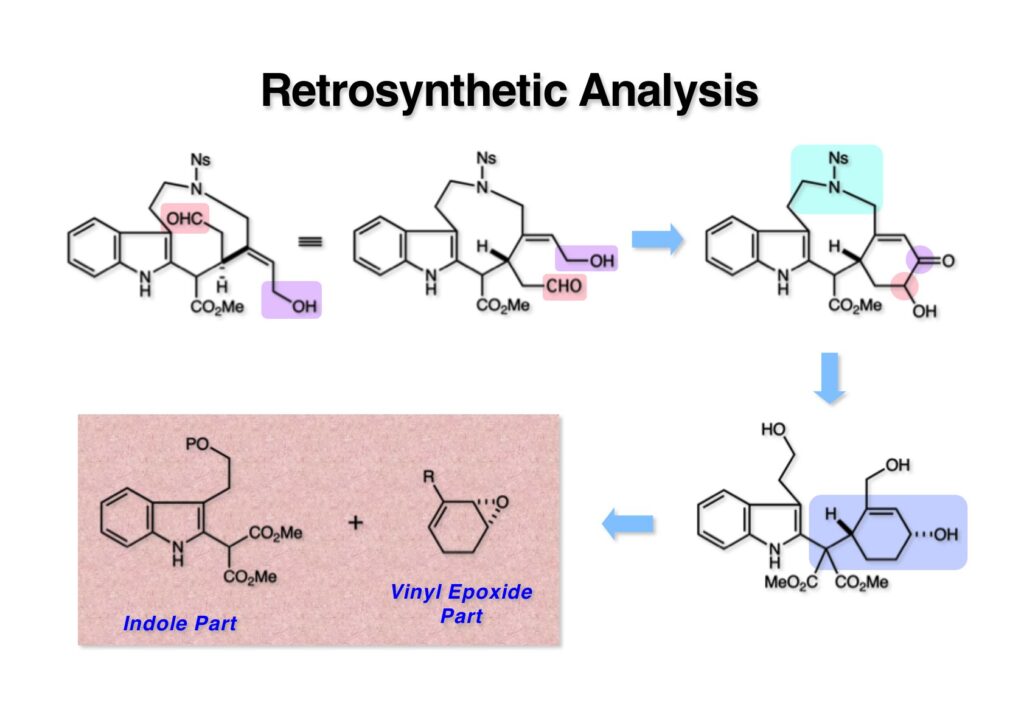
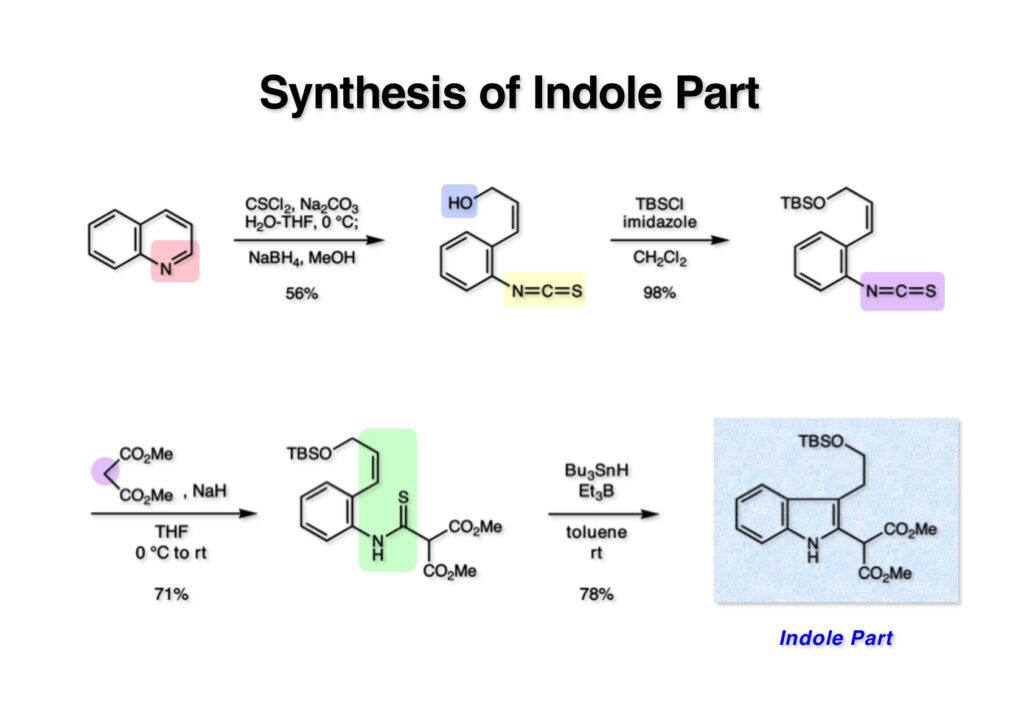
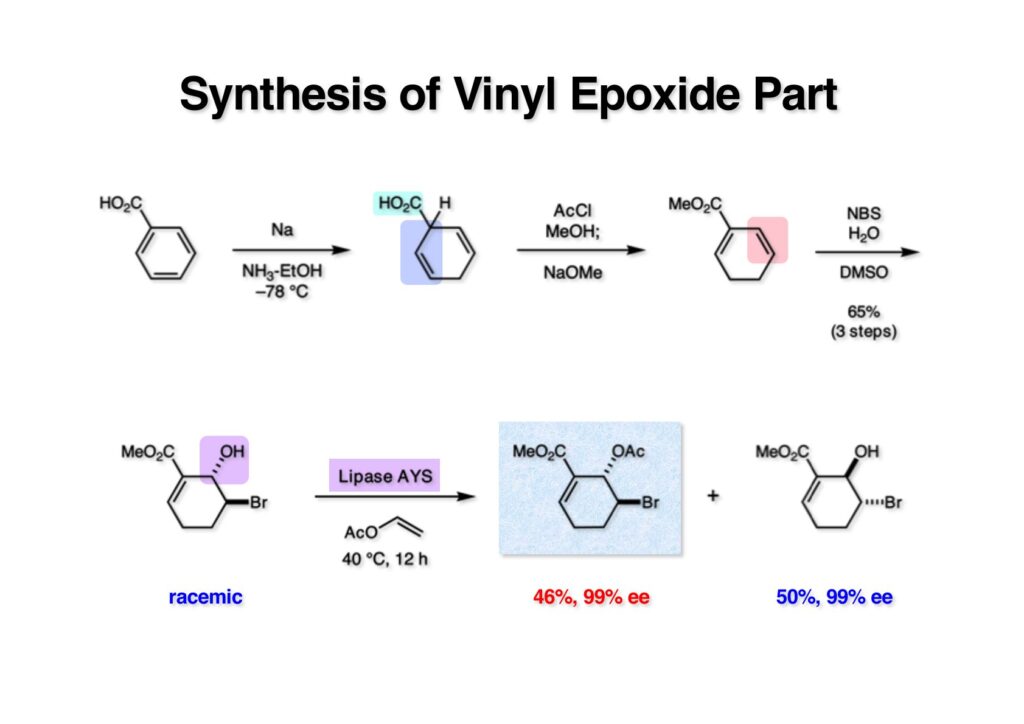
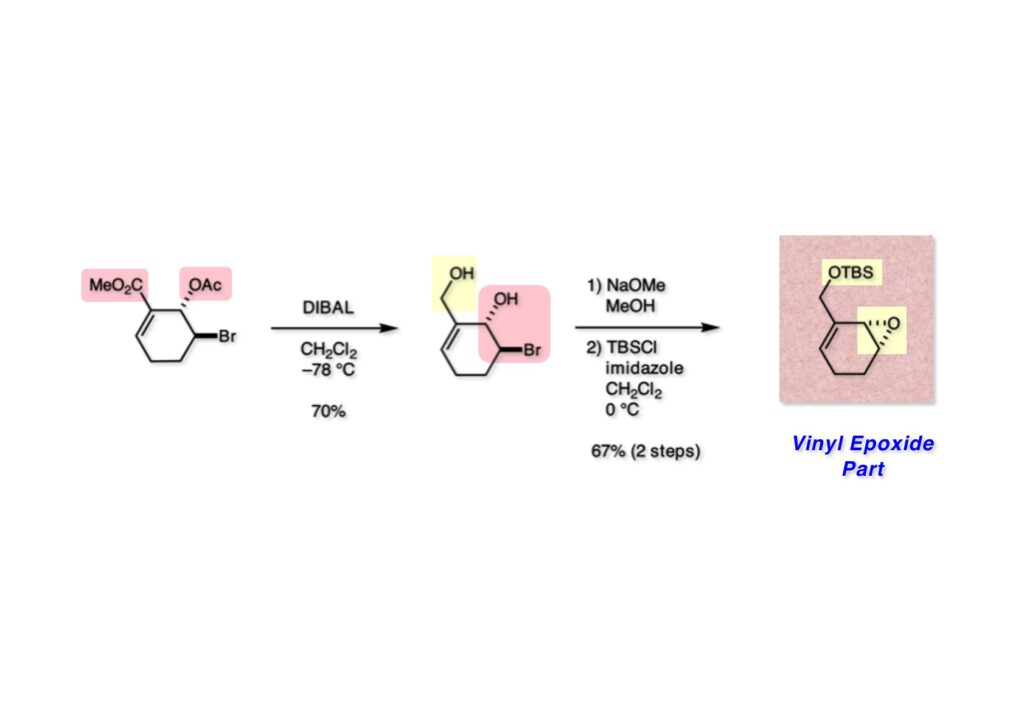
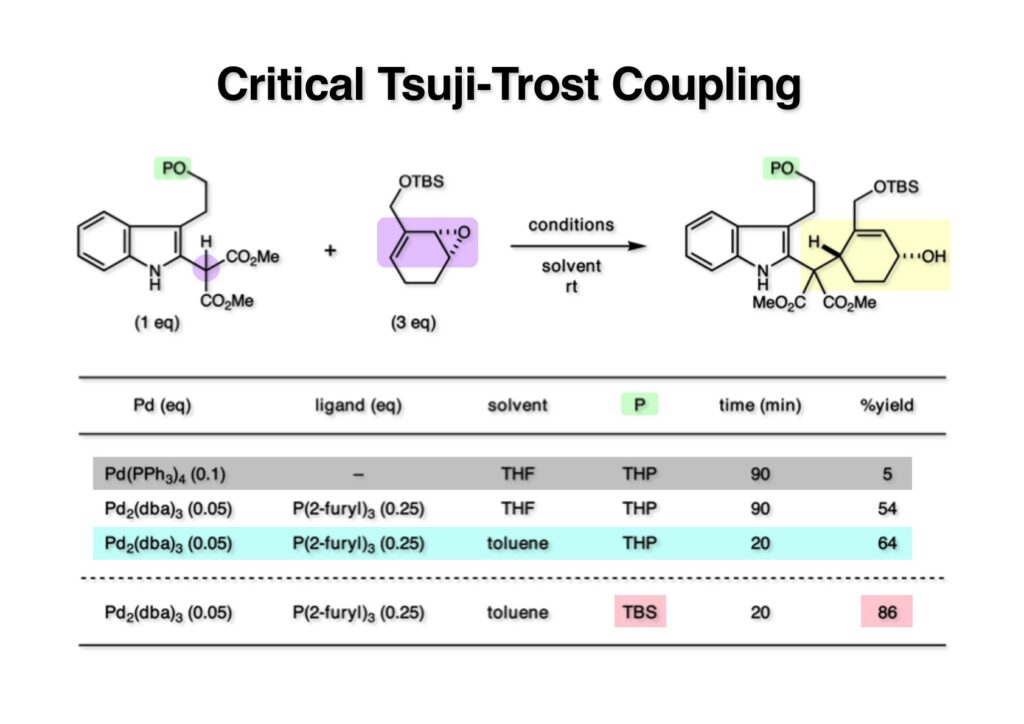
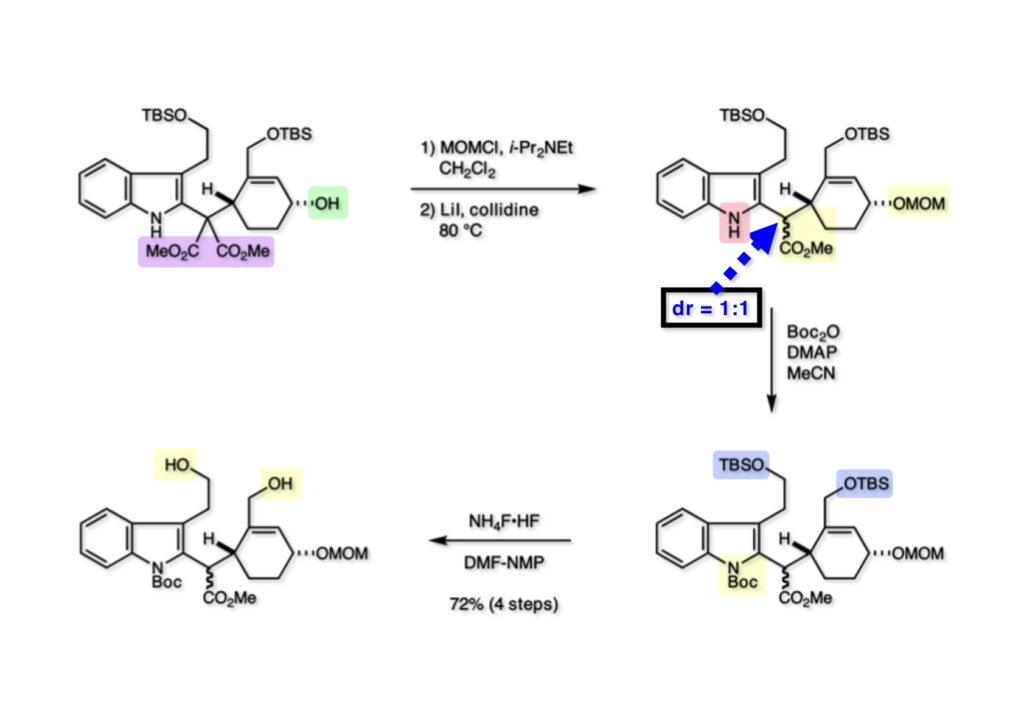
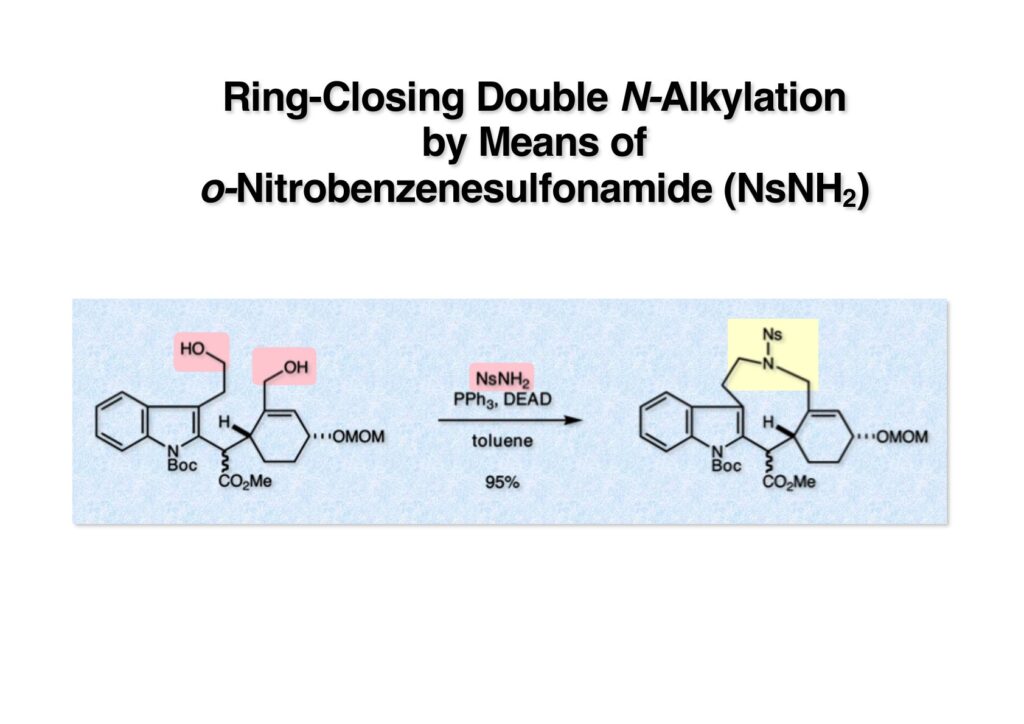
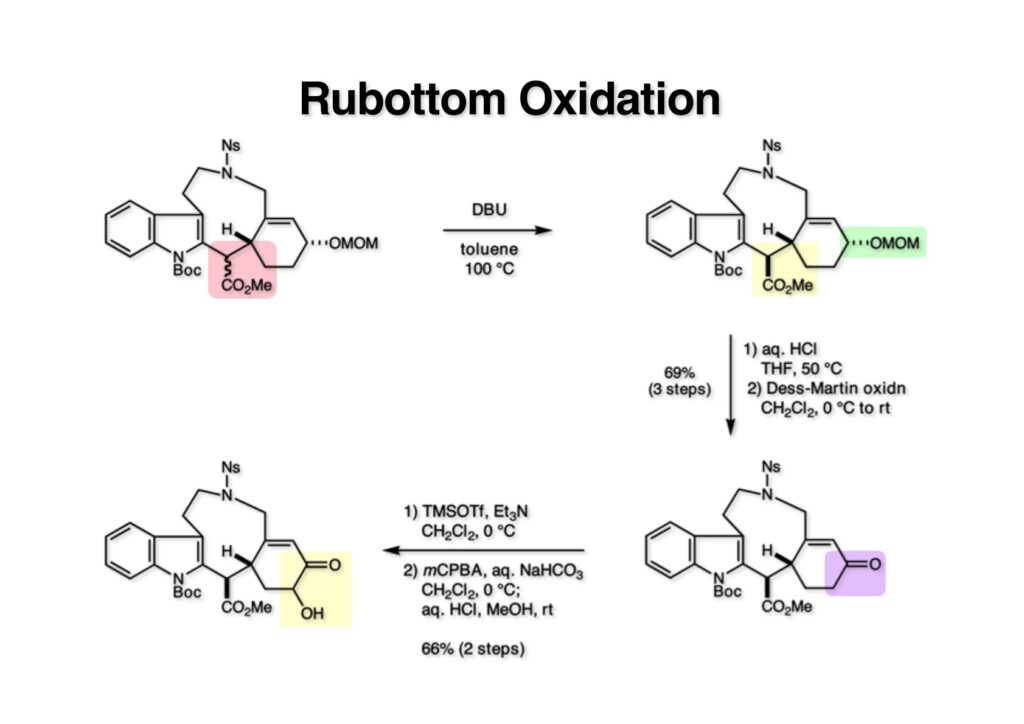
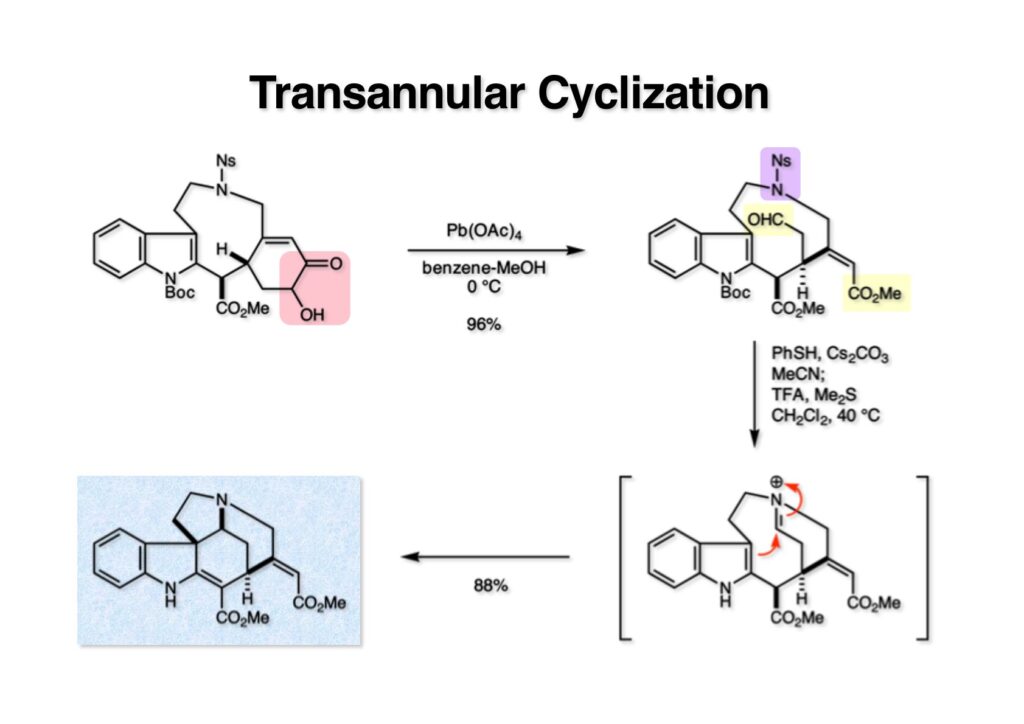
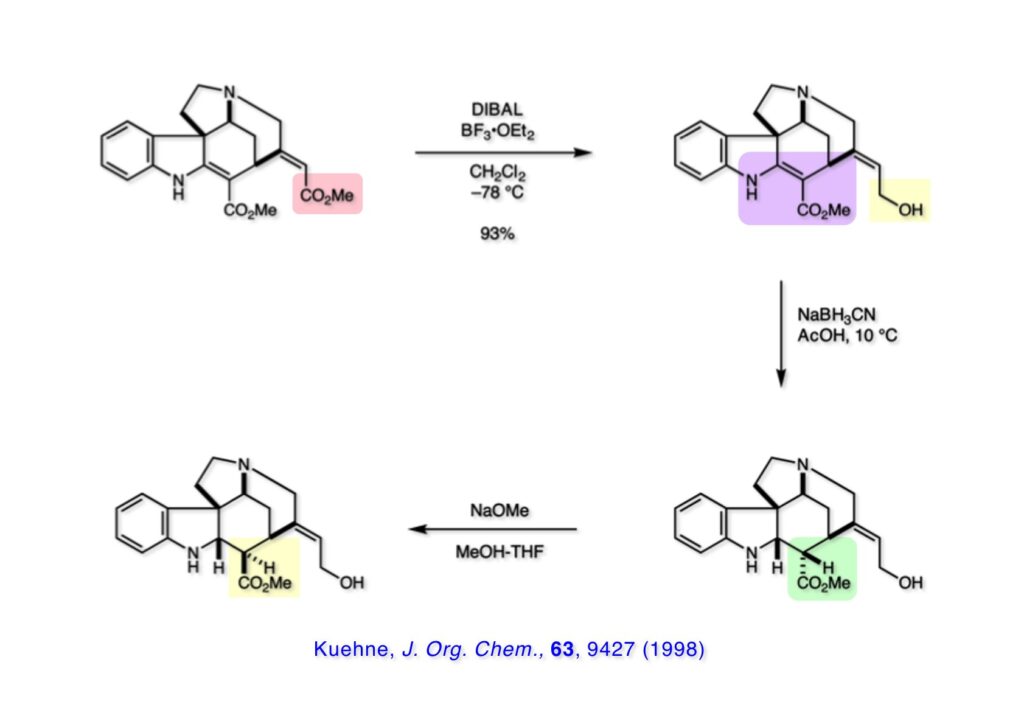
ストリキニーネの全合成はたまたま第二世代インドール合成法を開発し、ノシル化学とのコンビネーションで面白い研究になりそうだと思って始めた。勿論、有名な化合物であるし、構造的にも面白いと思ったが、主目的としては当研究室で開発した反応で独自の合成ルートを見出すことにあった。ちょうどleustroducsin Bの全合成の助太刀を終えた徳山さん配下の鏑木洋介君(現エーザイ)の手が空いたので彼一人に全合成を任せることにした。
“Total Synthesis of (–)-Strychnine,” Y. Kaburagi, H. Tokuyama, and T. Fukuyama, J. Am. Chem. Soc., 126, 10246 (2004)