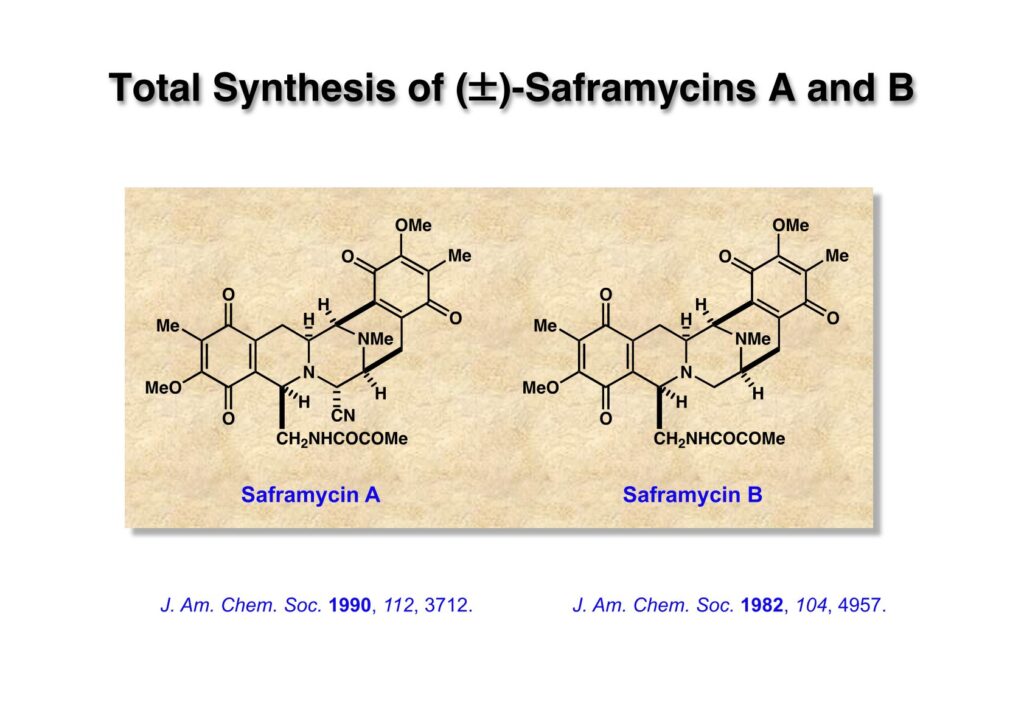
Saframycin Bはsaframycin Aなどと共に放線菌から単離された珍しいビスキノン型のイソキノリン二量体である。後に三共株式会社の副社長になった杉村征夫さんから構造情報を教えてもらった。杉村さんは阪大薬の北泰行先生と同じ頃にMITのBüchi研でポスドクをやられていて、自動車の運転も教えてもらったけれど授業料は払っていない。TFが院生の時にBüchi研で黒板を使った講演をしてほしいと頼まれ、最前列にBüchi先生が座られる中で博士論文の内容を開陳した。Büchi先生には大変気に入られ、後に就職活動をした時に推薦状を書いて頂いたし、昇進時にもその都度親切な推薦状を書いて頂いた。Saframycin Aは難易度が高かったので、まずは一番簡単なsaframycin Bの全合成を大学院1年のRick Sachlebenと開始した。Rickは頭が良く、やる気もある学生だったが、少々荒っぽいところが玉に瑕で、私が何かで注意したらフラストレーションが限界を超えたようで、実験台のキャビネットを思い切り蹴ったら戸がぶっ壊れた思い出がある。
“Stereocontrolled Total Synthesis of dl-Saframycin B,” T. Fukuyama and R. A. Sachleben, J. Am. Chem. Soc., 104, 4957-4958 (1982).
“Total Synthesis of (±)-Saframycin A,” T. Fukuyama, L.-H. Yang, K. L. Ajeck, and R. A. Sachleben, J. Am. Chem. Soc., 112, 3712 (1990).
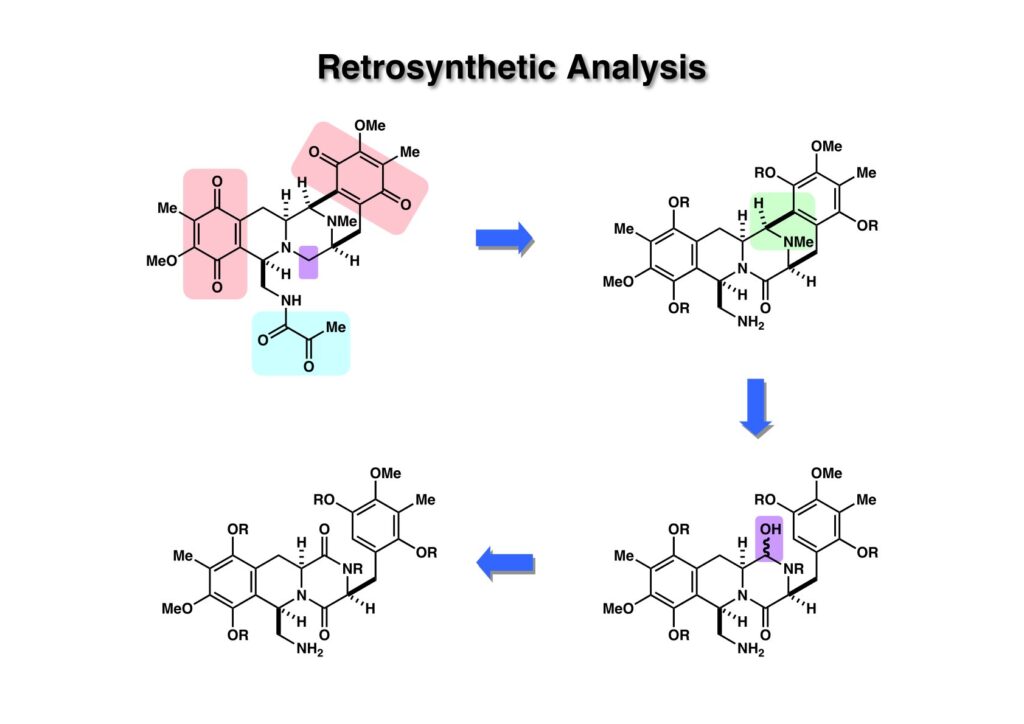 キノンは酸性および中性条件では比較的安定だが、塩基性条件下では不可脱離反応が起きやすく扱いにくい環である。従って、まず芳香環として保護しておいて、合成の最終段階に近いところで酸化によりキノンを構築するのが常道である。Pyruvoyl基も外しておくとアミン体 (1-2) に導かれる。 (1-2) のビシクロ [3.3.1] 構造を構築するには、Mannichi反応もしくはより強力なaxyliminium塩を形成して電子豊富な芳香環と反応させれば良いので (2-2) が前駆体と考えられる。ヘミアミナールを酸化すれば、ジケトピペラジン (2-1) となる。
キノンは酸性および中性条件では比較的安定だが、塩基性条件下では不可脱離反応が起きやすく扱いにくい環である。従って、まず芳香環として保護しておいて、合成の最終段階に近いところで酸化によりキノンを構築するのが常道である。Pyruvoyl基も外しておくとアミン体 (1-2) に導かれる。 (1-2) のビシクロ [3.3.1] 構造を構築するには、Mannichi反応もしくはより強力なaxyliminium塩を形成して電子豊富な芳香環と反応させれば良いので (2-2) が前駆体と考えられる。ヘミアミナールを酸化すれば、ジケトピペラジン (2-1) となる。
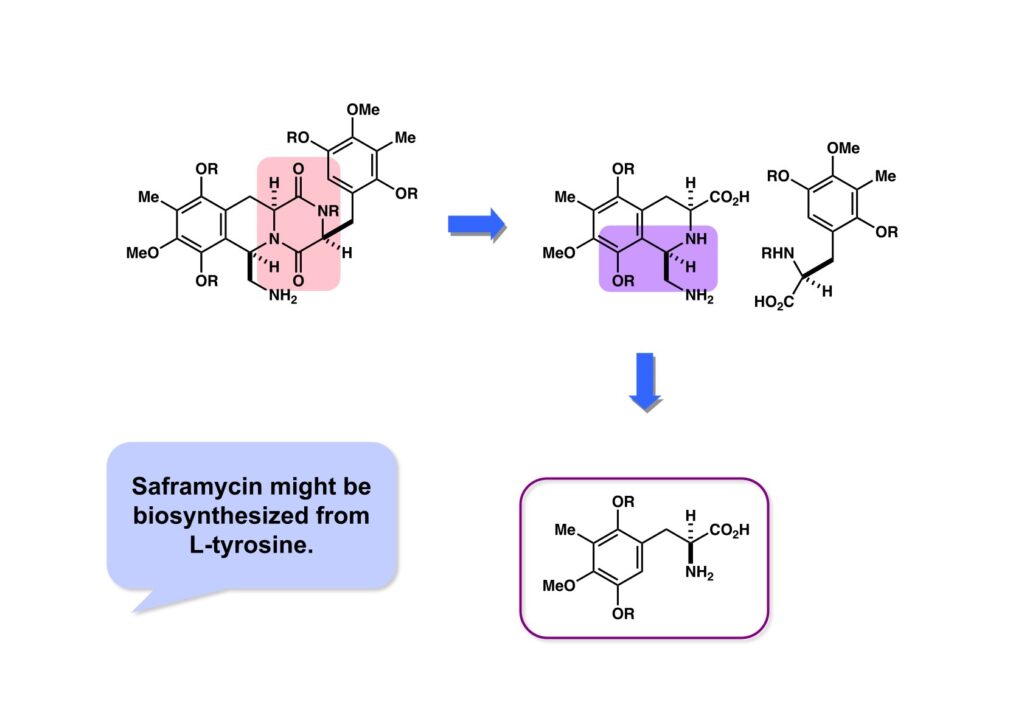 ジケトピペラジン (1-1) は2分子のアミノ酸が縮合した化合物なので加水分解して二つのアミノ酸. (1-2) と (1-3) に導ける。(1-2) はtetrahydroisoquinolineであるからMannich反応によってアミノ酸 (2-1) から容易に得られると考えられる。
ジケトピペラジン (1-1) は2分子のアミノ酸が縮合した化合物なので加水分解して二つのアミノ酸. (1-2) と (1-3) に導ける。(1-2) はtetrahydroisoquinolineであるからMannich反応によってアミノ酸 (2-1) から容易に得られると考えられる。
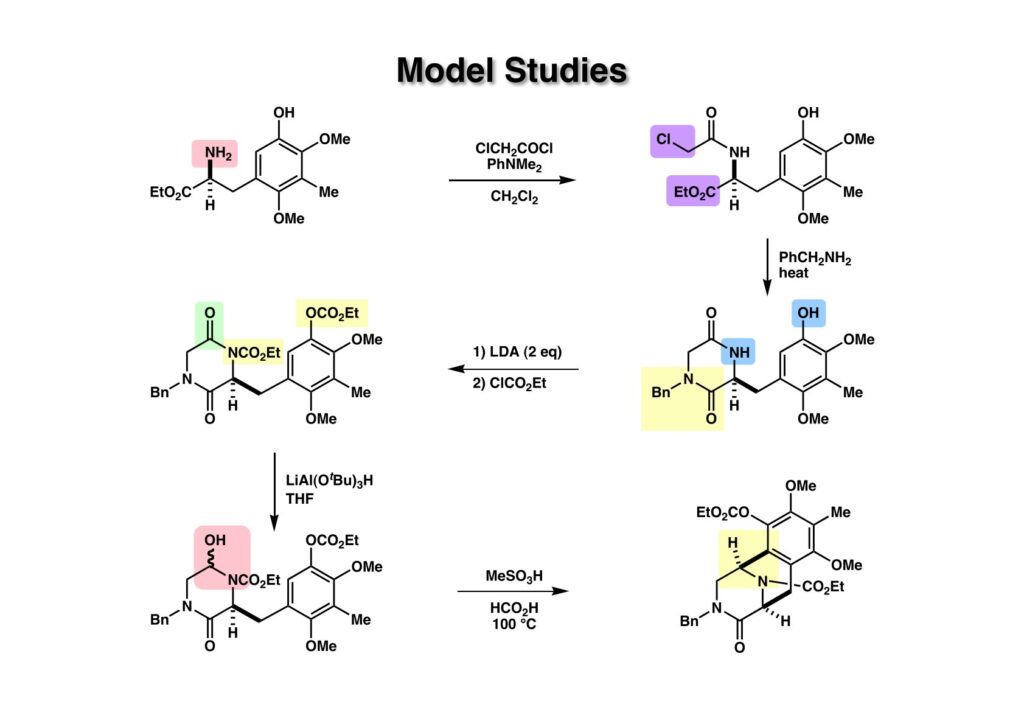 全合成を開始するにあたり、まずビシクロ体がどのような条件で構築できるかをモデル化合物を使って試してみた。出発物であるアミノ酸エステル (1-1) の合成は後述するが、アミンをClCH2COClでアシル化して (1-2) を得た後にベンジルアミンと加熱することでジケトピペラジン (2-2) が得られた。(2-2) を2当量のLDAで処理してClCO2Etを加えるとフェノールとラクタムがアシル化された (2-1) を得た。これを傘高くて反応性の比較的低いLiAl(OBu-t)3Hで還元することでヘミアミナール体 (3-1) が得られた。次にMeSO3Hを加えたギ酸中で加熱すると、望むビシクロ体 (3-2) がクリーンに得られた。
全合成を開始するにあたり、まずビシクロ体がどのような条件で構築できるかをモデル化合物を使って試してみた。出発物であるアミノ酸エステル (1-1) の合成は後述するが、アミンをClCH2COClでアシル化して (1-2) を得た後にベンジルアミンと加熱することでジケトピペラジン (2-2) が得られた。(2-2) を2当量のLDAで処理してClCO2Etを加えるとフェノールとラクタムがアシル化された (2-1) を得た。これを傘高くて反応性の比較的低いLiAl(OBu-t)3Hで還元することでヘミアミナール体 (3-1) が得られた。次にMeSO3Hを加えたギ酸中で加熱すると、望むビシクロ体 (3-2) がクリーンに得られた。
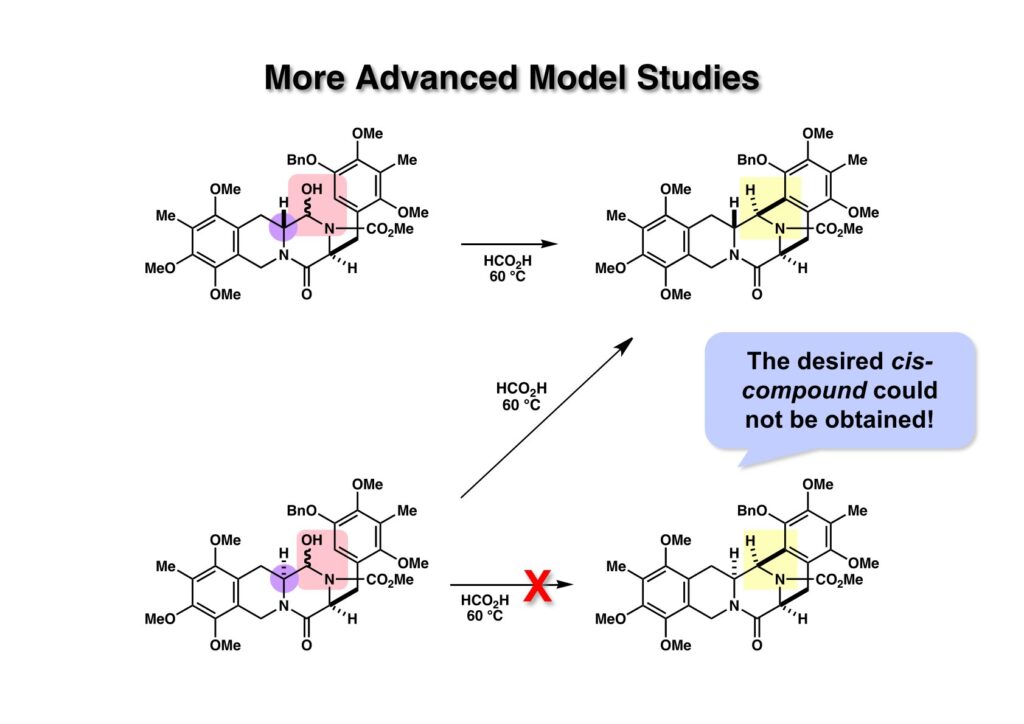 上述のモデル化合物では4位に置換基が入っていないので、さらに本番に近いモデル化合物を使って鍵反応が進行するかどうかを確かめた。cis-体 (2-1) とtrans-体 (1-1) を環化条件に付したところ同じ化合物 (1-2) が生成した。これは少々恐れていた結果で、trans-体が都合よくcis-体 (2-2) を与えるとは考えにくいので、確実にcis-体だと分かっている化合物について環化反応を行うことにした。
上述のモデル化合物では4位に置換基が入っていないので、さらに本番に近いモデル化合物を使って鍵反応が進行するかどうかを確かめた。cis-体 (2-1) とtrans-体 (1-1) を環化条件に付したところ同じ化合物 (1-2) が生成した。これは少々恐れていた結果で、trans-体が都合よくcis-体 (2-2) を与えるとは考えにくいので、確実にcis-体だと分かっている化合物について環化反応を行うことにした。
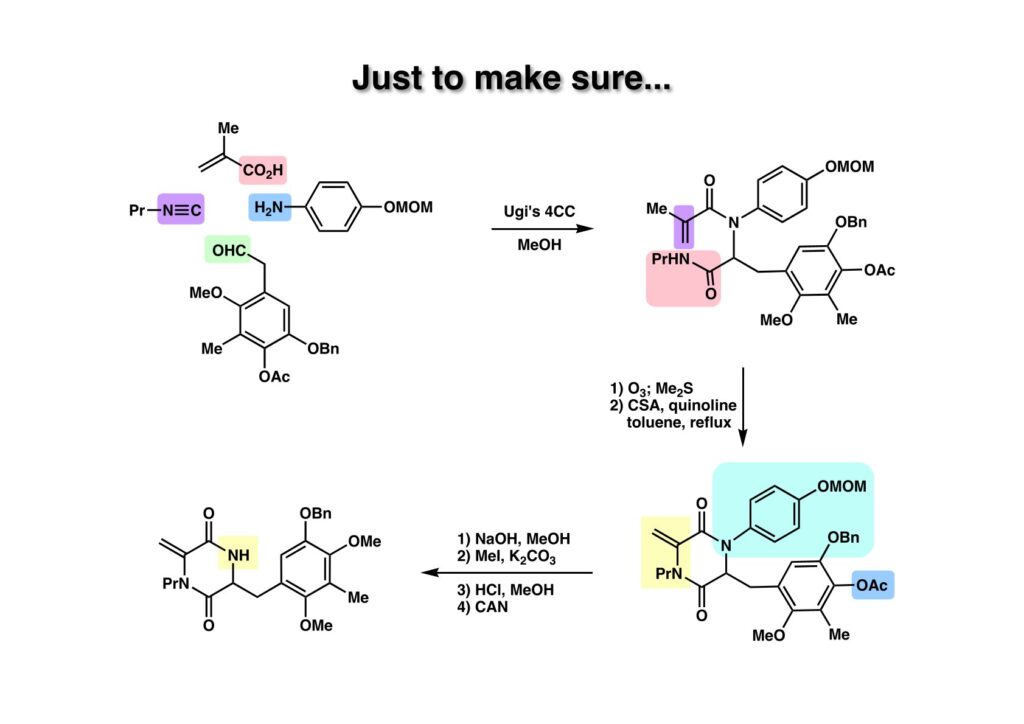 例のUgi反応を使って (1-2) を得た後、二重結合をオゾン分解し、CSAーキノリン存在下で加熱してジケトピペラジン (2-2) を得た。次にアセテートを加水分解して生じたフェノールをメチル化し、さらにMOM基を酸性条件で落としてからCAN酸化すると (2-1) が得られた。
例のUgi反応を使って (1-2) を得た後、二重結合をオゾン分解し、CSAーキノリン存在下で加熱してジケトピペラジン (2-2) を得た。次にアセテートを加水分解して生じたフェノールをメチル化し、さらにMOM基を酸性条件で落としてからCAN酸化すると (2-1) が得られた。
 (1-1) のようなジケトピペラジンを接触還元するとcis-体 (1-2) が得られることは文献既知であり、これを前述の条件で (2-2) に導いた。これを環化条件に付したところ、残念ながら望む環化体 (2-1) は得られなかった。ちょっとした立体障害でも期待する環化が起きなかったので、何とかしてこの問題を解決する必要に迫られた。
(1-1) のようなジケトピペラジンを接触還元するとcis-体 (1-2) が得られることは文献既知であり、これを前述の条件で (2-2) に導いた。これを環化条件に付したところ、残念ながら望む環化体 (2-1) は得られなかった。ちょっとした立体障害でも期待する環化が起きなかったので、何とかしてこの問題を解決する必要に迫られた。
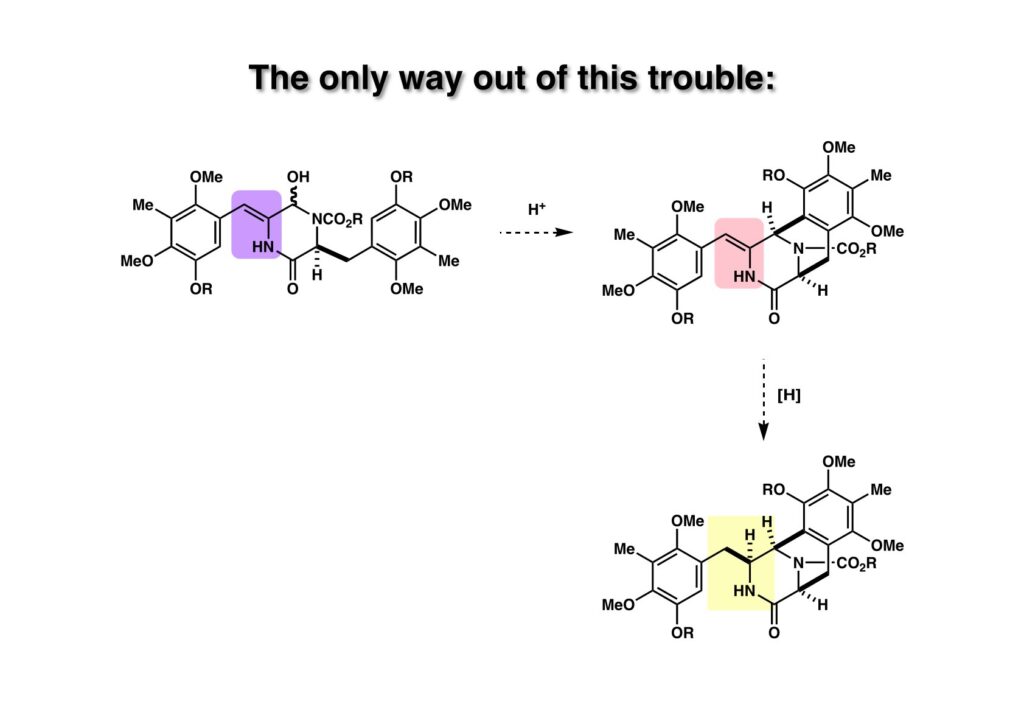 解決策はそれほど難しいことではなく、ビシクロ[3.3.1]体のendo側に側鎖を配向させるには (1-2) のような化合物の二重結合を空いているexo側から接触還元すれば可能であると考えた。
解決策はそれほど難しいことではなく、ビシクロ[3.3.1]体のendo側に側鎖を配向させるには (1-2) のような化合物の二重結合を空いているexo側から接触還元すれば可能であると考えた。
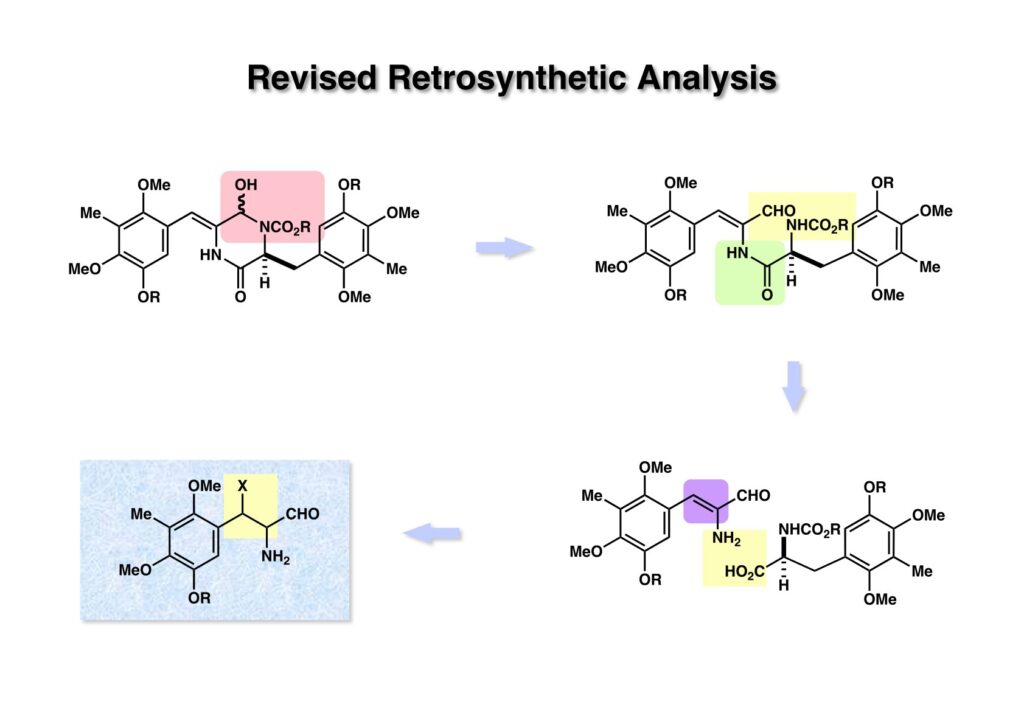 ヘミアミナール体 (1-1) のCN結合を開裂させるとα,β-不飽和アルデヒド (1-2) となる。ここでアミド結合を切ると実際には存在困難なα-amino-α,β-unsaturated aldehyde (2-2) とアミノ酸 (2-3) に導かれる。このアルデヒドに脱離基となるものを付加させておけば、(Nが保護されれば)比較的安定な (2-1) を合成すれば良いということになる。
ヘミアミナール体 (1-1) のCN結合を開裂させるとα,β-不飽和アルデヒド (1-2) となる。ここでアミド結合を切ると実際には存在困難なα-amino-α,β-unsaturated aldehyde (2-2) とアミノ酸 (2-3) に導かれる。このアルデヒドに脱離基となるものを付加させておけば、(Nが保護されれば)比較的安定な (2-1) を合成すれば良いということになる。
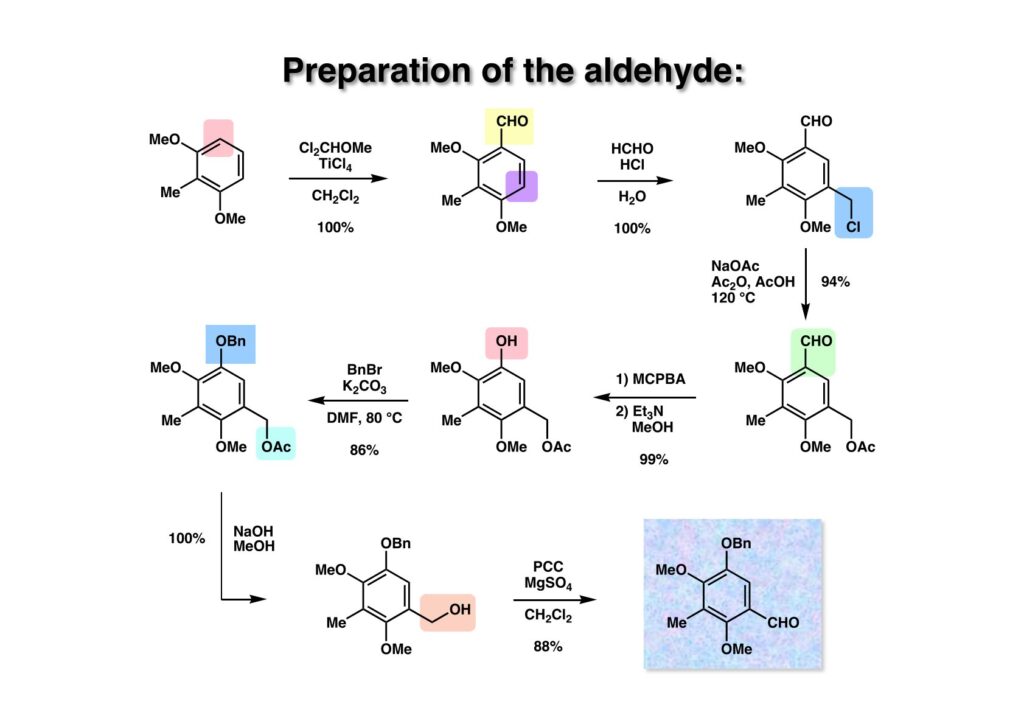 市販の2,6-dimethoxytoluene (1-1) をCl2CHOMe-TiCl4を使ってFriedel-Crafts反応を行いアルデヒド (1-2) を得た。この反応は試薬を加え終われば完結し、あとは氷にぶっ込んで抽出すると定量的に生成物が得られるので、Vilsmeier-Haack反応とは比較にならない。次に古典的な芳香環クロロメチル化反応で (1-3) を得た。出発物とホルマリンを混ぜておいてHClガスをぶくぶく通じるだけである。このクロライドをNaOAc、無水酢酸と酢酸中で加熱することでアセテート (2-3) に変換した。パラ位かオルト位に電子供与基がある芳香族アルデヒドは過酸酸化でギ酸エステルを与える。例えばベンズアルデヒドの過酸酸化では安息香酸が生ずるが、アニスアルデヒドではformateが生成する。 (2-3) は電子豊富なアルデヒドなので簡単にformateを与え、これをメタノール中でEt3Nで処理することでフェノール (2-2) が得られる。この手法は methyl formateが生成するのでエバポレータで飛ばすだけで生成物が得られて便利である。下手にNaOHなどを使うと抽出操作が必要になり手間がかかるだけである。実験する時には如何にして手間を省くかを常に考えておかなければならない。フェノール (2-2) をベンジル化して得られた (2-1) のアセテートを加水分解してアルコール (3-1) を得た後にPCC酸化でアルデヒド (3-2) を得た。
市販の2,6-dimethoxytoluene (1-1) をCl2CHOMe-TiCl4を使ってFriedel-Crafts反応を行いアルデヒド (1-2) を得た。この反応は試薬を加え終われば完結し、あとは氷にぶっ込んで抽出すると定量的に生成物が得られるので、Vilsmeier-Haack反応とは比較にならない。次に古典的な芳香環クロロメチル化反応で (1-3) を得た。出発物とホルマリンを混ぜておいてHClガスをぶくぶく通じるだけである。このクロライドをNaOAc、無水酢酸と酢酸中で加熱することでアセテート (2-3) に変換した。パラ位かオルト位に電子供与基がある芳香族アルデヒドは過酸酸化でギ酸エステルを与える。例えばベンズアルデヒドの過酸酸化では安息香酸が生ずるが、アニスアルデヒドではformateが生成する。 (2-3) は電子豊富なアルデヒドなので簡単にformateを与え、これをメタノール中でEt3Nで処理することでフェノール (2-2) が得られる。この手法は methyl formateが生成するのでエバポレータで飛ばすだけで生成物が得られて便利である。下手にNaOHなどを使うと抽出操作が必要になり手間がかかるだけである。実験する時には如何にして手間を省くかを常に考えておかなければならない。フェノール (2-2) をベンジル化して得られた (2-1) のアセテートを加水分解してアルコール (3-1) を得た後にPCC酸化でアルデヒド (3-2) を得た。
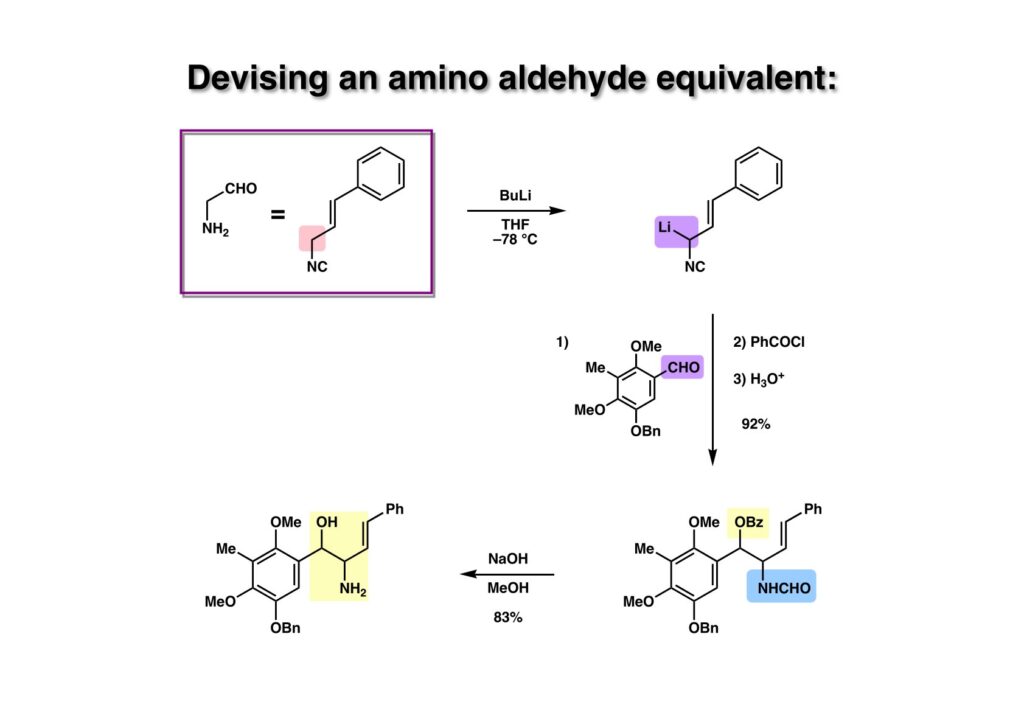 Ugi反応に興味を持ってイソニトリルの化学を調べていた時に、Ulrich Schöllkopf教授やDieter Hoppe教授のAngewandte Chemieの総説は読み込んでいた。イソニトリルのα位プロトンが酸性を持つと言うことで、これを利用すればアミノアルデヒドが簡単に得られると直感した。Cinnamyl isocyanide (1-2) を低温下でBuLiで脱プロトン化し、先ほど合成したアルデヒド (2-) を加えて昇温したところ、所望のアルコールは得られず、イソニトリルに付加したオキサゾリンが生成してしまった。この環化反応は低温下では進行しないことが分かったためPhCOClを加えてベンゾエートにし、単離することなく反応液に塩酸を加えてイソニトリルを水和してホルムアミド (3-2) に変換した。次にNaOHを用いてホルムアミドとベンゾエートを加メタノール分解して望むアミノアルコール (3-1) を得た。
Ugi反応に興味を持ってイソニトリルの化学を調べていた時に、Ulrich Schöllkopf教授やDieter Hoppe教授のAngewandte Chemieの総説は読み込んでいた。イソニトリルのα位プロトンが酸性を持つと言うことで、これを利用すればアミノアルデヒドが簡単に得られると直感した。Cinnamyl isocyanide (1-2) を低温下でBuLiで脱プロトン化し、先ほど合成したアルデヒド (2-) を加えて昇温したところ、所望のアルコールは得られず、イソニトリルに付加したオキサゾリンが生成してしまった。この環化反応は低温下では進行しないことが分かったためPhCOClを加えてベンゾエートにし、単離することなく反応液に塩酸を加えてイソニトリルを水和してホルムアミド (3-2) に変換した。次にNaOHを用いてホルムアミドとベンゾエートを加メタノール分解して望むアミノアルコール (3-1) を得た。
 アミノ酸 (2-1) の合成は前述のアルデヒド (1-1) とethyl isocyanoacetate (1-2) からSchöllkopfのアミノ酸合成を用いて行なった。両者の縮合はKHを使うことで室温で進行し、α,β-不飽和エステル (1-3) が得られた。接触還元で二重結合を飽和させ、脱落したベンジル基をかけ直して (2-2) に変換した。次にホルミル基を酸性で除去してからCbzClでアシル化、さらにエステルを加水分解してカルボン酸 (2-1) が得られた。
アミノ酸 (2-1) の合成は前述のアルデヒド (1-1) とethyl isocyanoacetate (1-2) からSchöllkopfのアミノ酸合成を用いて行なった。両者の縮合はKHを使うことで室温で進行し、α,β-不飽和エステル (1-3) が得られた。接触還元で二重結合を飽和させ、脱落したベンジル基をかけ直して (2-2) に変換した。次にホルミル基を酸性で除去してからCbzClでアシル化、さらにエステルを加水分解してカルボン酸 (2-1) が得られた。
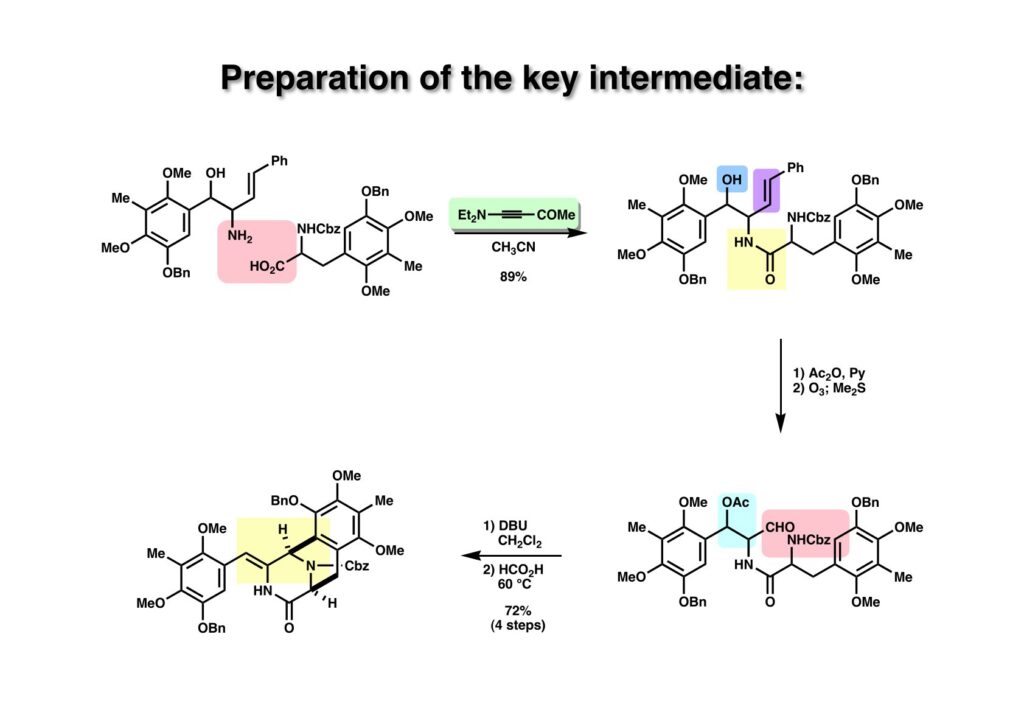 アミン (1-1) とカルボン酸 (1-2) の縮合はynamine誘導体 (1-3) を用いた。これはドイツの化学者が報告していたが、反応機構が面白いので使ってみたかったと言うのが理由である。得られた (1-4) の水酸基をアセチル化してから二重結合をオゾン分解することでアルデヒド (2-2) に導いた。次いで、DBU処理で酢酸を脱離させた後にギ酸中で加熱したところ、望む閉環反応が進行してビシクロ体 (2-1) を得ることができた。ギ酸を加えると溶液が血のように真っ赤になり、血の色が消えると反応が終了していた。あの真っ赤な化合物の正体は何だったのだろう。
アミン (1-1) とカルボン酸 (1-2) の縮合はynamine誘導体 (1-3) を用いた。これはドイツの化学者が報告していたが、反応機構が面白いので使ってみたかったと言うのが理由である。得られた (1-4) の水酸基をアセチル化してから二重結合をオゾン分解することでアルデヒド (2-2) に導いた。次いで、DBU処理で酢酸を脱離させた後にギ酸中で加熱したところ、望む閉環反応が進行してビシクロ体 (2-1) を得ることができた。ギ酸を加えると溶液が血のように真っ赤になり、血の色が消えると反応が終了していた。あの真っ赤な化合物の正体は何だったのだろう。
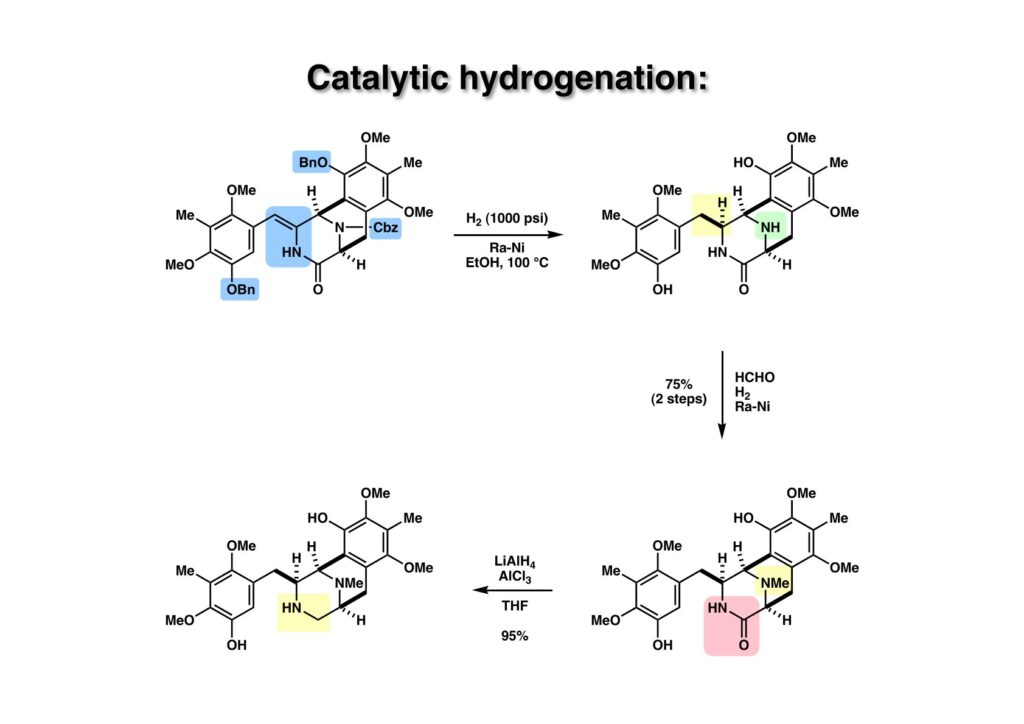 肝心な二重結合の接触還元はパラジウム触媒も白金触媒も歯が立たず、外れるのはベンジル基だけという結果だった。Raneyニッケルは高温高圧で威力を発揮する触媒で、ここでも頼りにはなったが、ステンレス製の反応容器で水素圧は1000 psi、温度は100度をかけてやっと還元が終了した (1-2)。ついでにアミンをメチル化しようと、ホルマリンを加えて水素添加を続行したところ目的物 (2-2) が得られた。ラクタム (2-2) のアミン (2-1) への還元は当初LiAlH4を用いたが低収率に終わった。そこでLiAlH4とAlCl3で水素化アルミニウム (alane) を調製して還元に用いたところ高収率でアミン (2-1) が得られた。以後はラクタムの還元は水素化アルミニウムを使うことにした。
肝心な二重結合の接触還元はパラジウム触媒も白金触媒も歯が立たず、外れるのはベンジル基だけという結果だった。Raneyニッケルは高温高圧で威力を発揮する触媒で、ここでも頼りにはなったが、ステンレス製の反応容器で水素圧は1000 psi、温度は100度をかけてやっと還元が終了した (1-2)。ついでにアミンをメチル化しようと、ホルマリンを加えて水素添加を続行したところ目的物 (2-2) が得られた。ラクタム (2-2) のアミン (2-1) への還元は当初LiAlH4を用いたが低収率に終わった。そこでLiAlH4とAlCl3で水素化アルミニウム (alane) を調製して還元に用いたところ高収率でアミン (2-1) が得られた。以後はラクタムの還元は水素化アルミニウムを使うことにした。
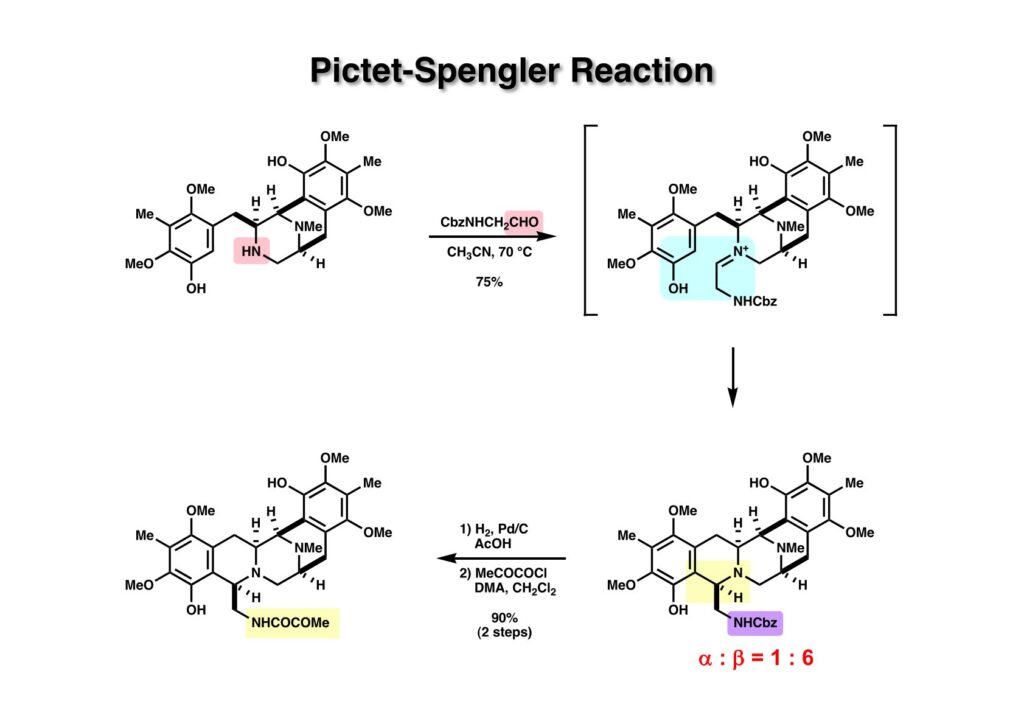 次に重要となる反応は (1-1) のPictet-Spengler反応で、相方がフェノールの場合に亀谷哲治先生はフェノール環化と呼ばれていた。アセトニトリル中でアルデヒド (1-2) と加熱すると、おそらく立体反発を避けたE-体のイミニウム塩 (1-3) が優先的に生成し、β-面から反応することによってβ-体 (2-2) が主生成物として得られたと考えられる。次にCbz基を還元的に除去してからpyruvoyl基を導入して (2-1) を得た。
次に重要となる反応は (1-1) のPictet-Spengler反応で、相方がフェノールの場合に亀谷哲治先生はフェノール環化と呼ばれていた。アセトニトリル中でアルデヒド (1-2) と加熱すると、おそらく立体反発を避けたE-体のイミニウム塩 (1-3) が優先的に生成し、β-面から反応することによってβ-体 (2-2) が主生成物として得られたと考えられる。次にCbz基を還元的に除去してからpyruvoyl基を導入して (2-1) を得た。
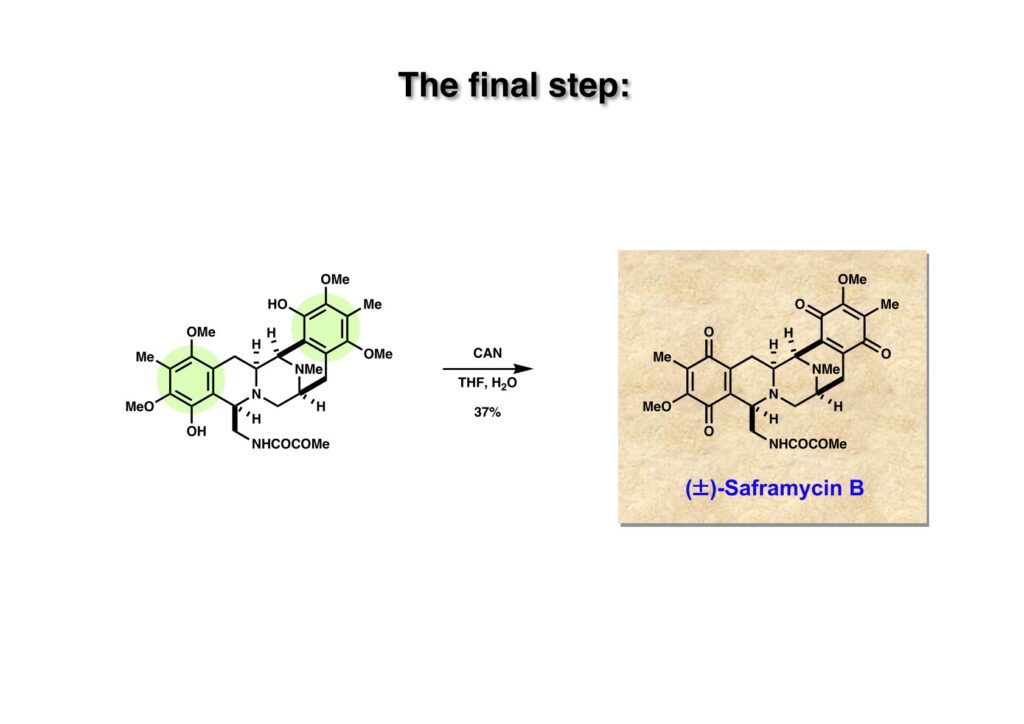 最終段階のフェノール (1-1) の酸化にはCANを用いたが、おそらくここもDDQを使った方が収率は高かったと思う。ひとまずsaframycin Bの最初の全合成はこれで完成した。
最終段階のフェノール (1-1) の酸化にはCANを用いたが、おそらくここもDDQを使った方が収率は高かったと思う。ひとまずsaframycin Bの最初の全合成はこれで完成した。
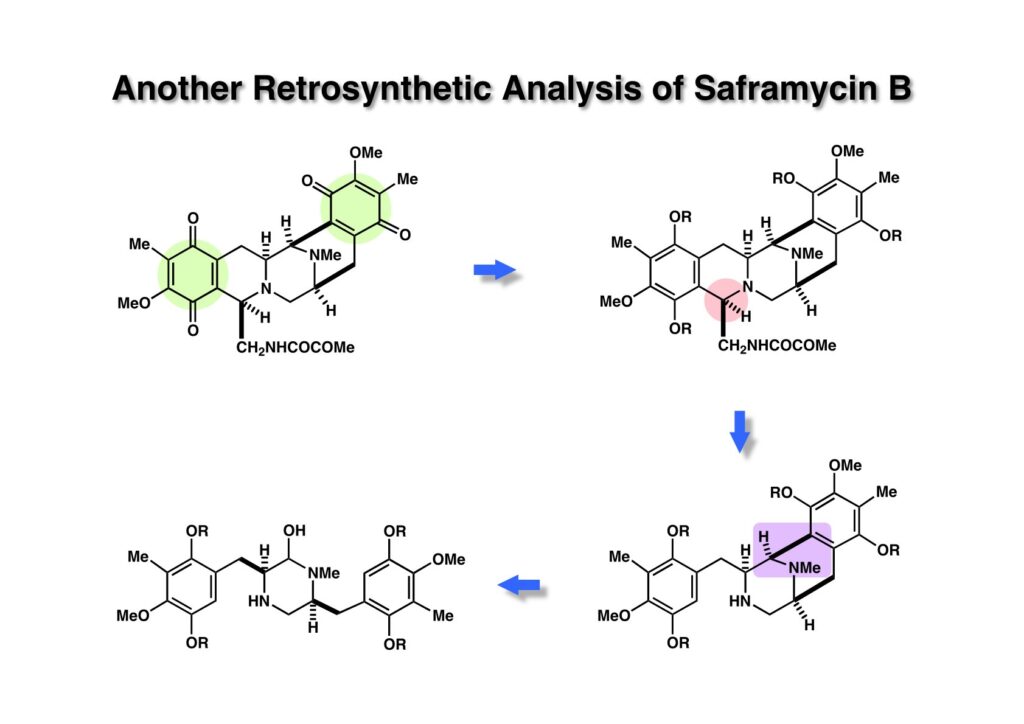 Saframycin Bの全合成は終わったが、抗腫瘍活性を示すsaframycin Aの全合成をやりたいと思い、逆合成解析を行なってみた。ここでは対称形のジケトピペラジンを非対称化することに興味を覚えた。単純化するためにsaframycin Bの逆合成解析をここでは示す。(1-1) のキノンを芳香環として保護した (1-2) の逆Pictet-Spengler反応で (2-2) に変換し、ここで逆Mannich反応で (2-1) へと導く。
Saframycin Bの全合成は終わったが、抗腫瘍活性を示すsaframycin Aの全合成をやりたいと思い、逆合成解析を行なってみた。ここでは対称形のジケトピペラジンを非対称化することに興味を覚えた。単純化するためにsaframycin Bの逆合成解析をここでは示す。(1-1) のキノンを芳香環として保護した (1-2) の逆Pictet-Spengler反応で (2-2) に変換し、ここで逆Mannich反応で (2-1) へと導く。
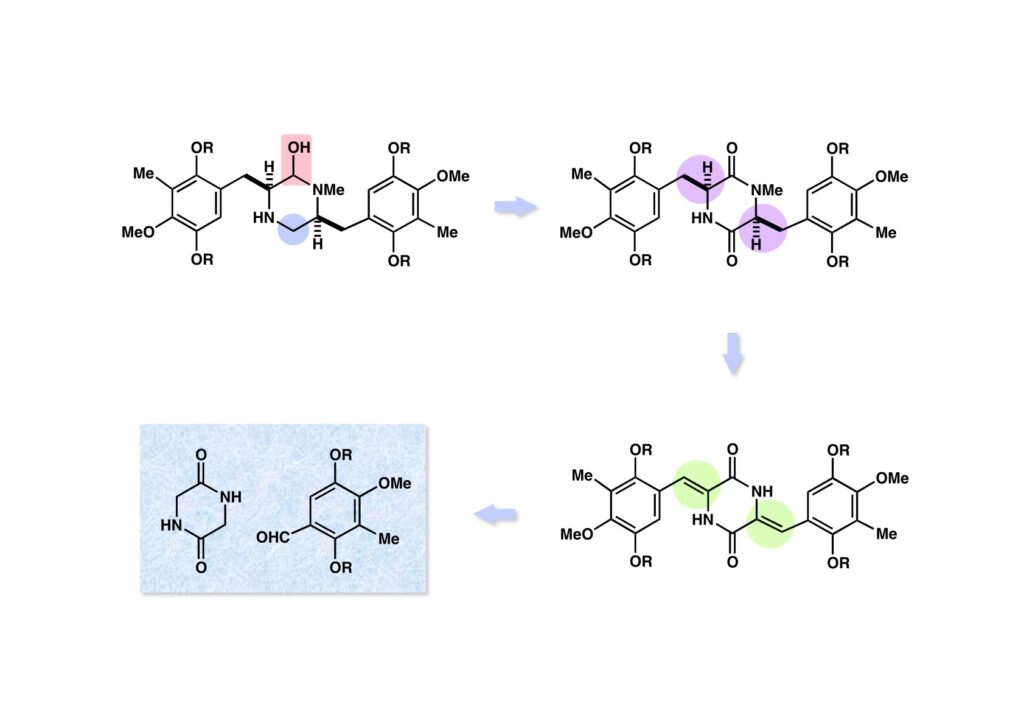 ヘミアミナール (1-1) の酸化とNHアミンの横にもカルボニル基を導入することでジケトピペラジン (1-2) に変換し、さらに飽和な側鎖をオレフィン (2-3) にすれば、ジケトピペラジン (2-1) とアルデヒド (2-2) の縮合反応に単純化することが可能になる。この逆合成的考察はハーバード大のポスドク時代にWelch Foundationへの研究費申請用プロポーザルでantibiotic 593Aの逆合成を考えていた時に、イタリア人化学者(GallinaとLiberatori )の1974年のTetrahedronの論文を見たから思いついた。
ヘミアミナール (1-1) の酸化とNHアミンの横にもカルボニル基を導入することでジケトピペラジン (1-2) に変換し、さらに飽和な側鎖をオレフィン (2-3) にすれば、ジケトピペラジン (2-1) とアルデヒド (2-2) の縮合反応に単純化することが可能になる。この逆合成的考察はハーバード大のポスドク時代にWelch Foundationへの研究費申請用プロポーザルでantibiotic 593Aの逆合成を考えていた時に、イタリア人化学者(GallinaとLiberatori )の1974年のTetrahedronの論文を見たから思いついた。
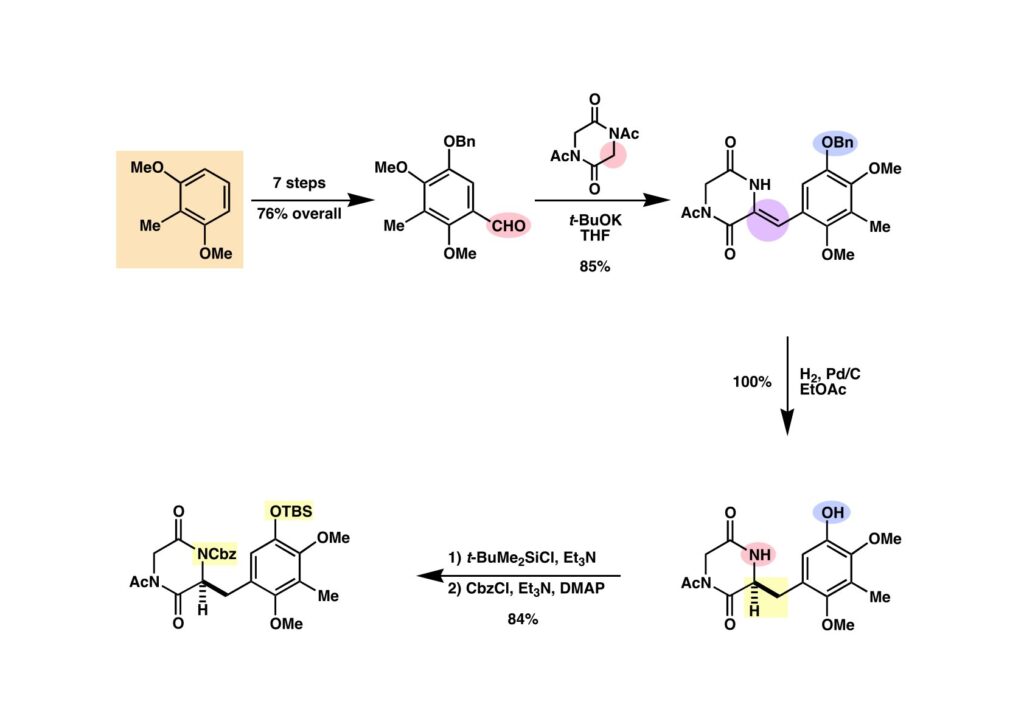 2,6-Dimethoxytoluene (1-1) から7段階で合成したアルデヒド (1-2) とN,N’-diacetylpiperazinedione (1-3) の混合物にt-BuOK/t-BuOHを加えると、GallinaとLiberatoriが報告したようにスムーズな縮合反応が進行して一箇所だけが反応した生成物 (1-4) が得られる。この反応はうまく出来ていて、アルデヒドに付加して生じたアルコキサイドが近隣のアセチル基を攻撃してアセテートになるがアルキル基が結合した炭素上の水素はイミドの酸性プロトンでありt-BuOKで容易に脱プロトン化されてアセテートが脱離する。そうなればイミドで活性化されたプロトンは消失し、それ以上の反応は起きないことになる。対称性を崩して非対称化する、と言うのはこの反応があったから可能となったと言える。(1-4) の二重結合を接触還元すると脱ベンジル化された化合物 (2-2) が得られる。このイミドのアセチル基は塩基性で簡単に外れてしまうので、フェノールの再ベンジル化は諦めてTBS化して保護した。次の縮合反応を行うためにラクタムのNHをCbz基を導入することでイミド (2-1) として活性化した。
2,6-Dimethoxytoluene (1-1) から7段階で合成したアルデヒド (1-2) とN,N’-diacetylpiperazinedione (1-3) の混合物にt-BuOK/t-BuOHを加えると、GallinaとLiberatoriが報告したようにスムーズな縮合反応が進行して一箇所だけが反応した生成物 (1-4) が得られる。この反応はうまく出来ていて、アルデヒドに付加して生じたアルコキサイドが近隣のアセチル基を攻撃してアセテートになるがアルキル基が結合した炭素上の水素はイミドの酸性プロトンでありt-BuOKで容易に脱プロトン化されてアセテートが脱離する。そうなればイミドで活性化されたプロトンは消失し、それ以上の反応は起きないことになる。対称性を崩して非対称化する、と言うのはこの反応があったから可能となったと言える。(1-4) の二重結合を接触還元すると脱ベンジル化された化合物 (2-2) が得られる。このイミドのアセチル基は塩基性で簡単に外れてしまうので、フェノールの再ベンジル化は諦めてTBS化して保護した。次の縮合反応を行うためにラクタムのNHをCbz基を導入することでイミド (2-1) として活性化した。
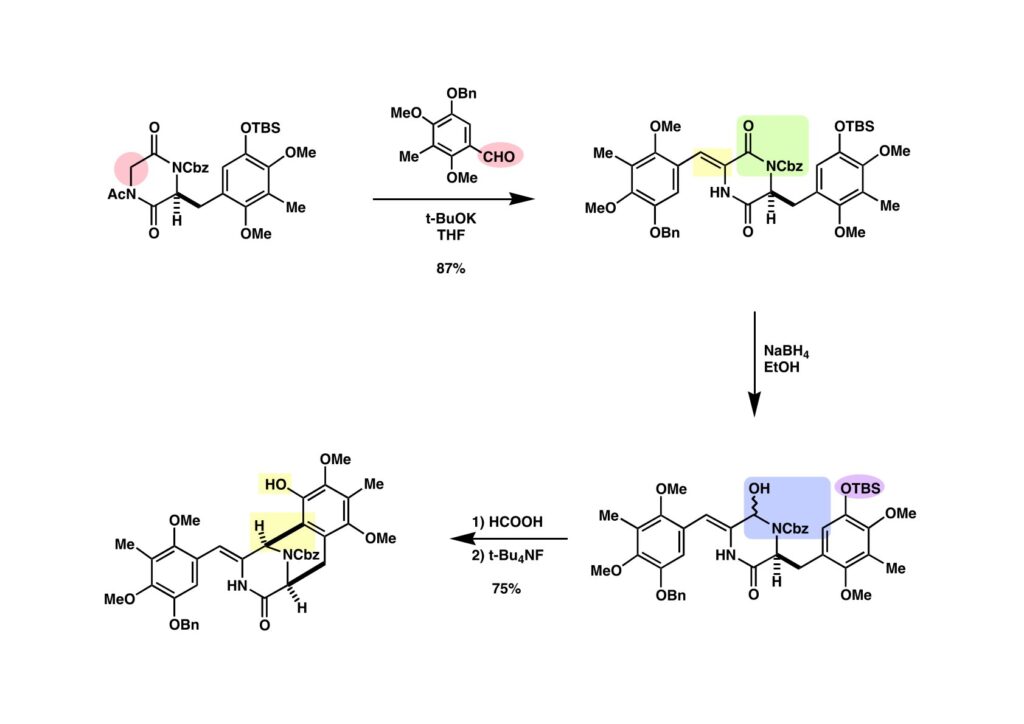 イミド (1-1) とアルデヒド (1-2) の混合物に再びt-BuOKを加えると、速度論的に有利なCH2側が脱プロトン化されて (1-2) が得られた。イミドカルボニル基をNaBH4で還元して (2-2)を得て、次いでギ酸処理することによりビシクロ体が生成した。ここでTBS基を落として (2-1) にしたのだが、その理由は二重結合の接触還元によってα体とβ体の混合物になってしまったからだ。(2-1) のモデルを組むとTBSO基がα面にまで突き出ていたことが分かる。
イミド (1-1) とアルデヒド (1-2) の混合物に再びt-BuOKを加えると、速度論的に有利なCH2側が脱プロトン化されて (1-2) が得られた。イミドカルボニル基をNaBH4で還元して (2-2)を得て、次いでギ酸処理することによりビシクロ体が生成した。ここでTBS基を落として (2-1) にしたのだが、その理由は二重結合の接触還元によってα体とβ体の混合物になってしまったからだ。(2-1) のモデルを組むとTBSO基がα面にまで突き出ていたことが分かる。
 (1-1) をRaneyニッケルで高圧下に水添すると定量的に目的物 (1-2) が得られた。このアミン (1-2) をreductive methylationすることでsaframycin Bの合成中間体を効率よく合成することができた。
(1-1) をRaneyニッケルで高圧下に水添すると定量的に目的物 (1-2) が得られた。このアミン (1-2) をreductive methylationすることでsaframycin Bの合成中間体を効率よく合成することができた。
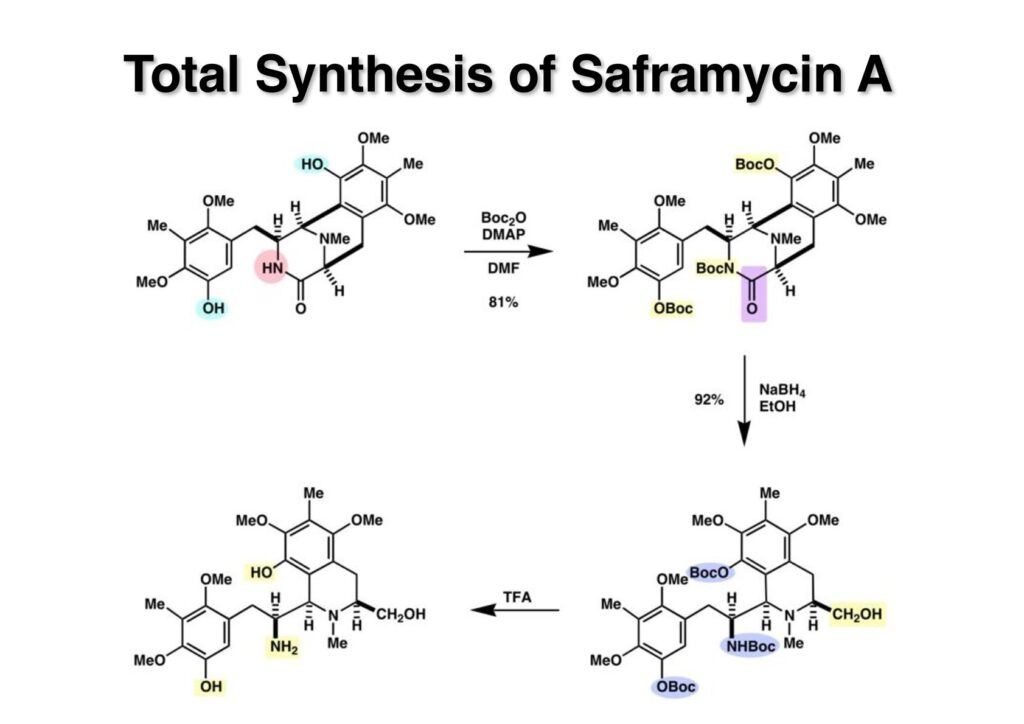 Saframycin Bとsaframycin Aの論文発表年に8年の間隔がある理由は、私自身のやる気が今一つ足らなかったのと、学生が良いところまで到達しながら、あと一息のプッシュが足らなかったのかもしれない。やる気が無いとはいえ、そう野ざらしにもしておけないので、mitomycin Cの全合成を終えたLihu Yangにこのプロジェクトを終わらせるように頼んだ。程なくLihuは合成を完成させたが、一番の鍵となったのはフェノール基2つとアミン存在下で一級アルコールのSwern酸化を成功させたところだろう。Saframycin Aを合成するためにはラクタム (1-1) のアミド結合を切ってPictet-Spengler反応を行わなければならない。(1-1) をBoc2OとDMAPで処理すると容易に二つのフェノールとラクタムNHにBoc基が導入された化合物 (1-2) が得られる。これをNaBH4で還元すると (2-2) を与え、更にTFA処理をすると3つのBoc基が落ちてアミノフェノール体 (2-1) に変換された。
Saframycin Bとsaframycin Aの論文発表年に8年の間隔がある理由は、私自身のやる気が今一つ足らなかったのと、学生が良いところまで到達しながら、あと一息のプッシュが足らなかったのかもしれない。やる気が無いとはいえ、そう野ざらしにもしておけないので、mitomycin Cの全合成を終えたLihu Yangにこのプロジェクトを終わらせるように頼んだ。程なくLihuは合成を完成させたが、一番の鍵となったのはフェノール基2つとアミン存在下で一級アルコールのSwern酸化を成功させたところだろう。Saframycin Aを合成するためにはラクタム (1-1) のアミド結合を切ってPictet-Spengler反応を行わなければならない。(1-1) をBoc2OとDMAPで処理すると容易に二つのフェノールとラクタムNHにBoc基が導入された化合物 (1-2) が得られる。これをNaBH4で還元すると (2-2) を与え、更にTFA処理をすると3つのBoc基が落ちてアミノフェノール体 (2-1) に変換された。
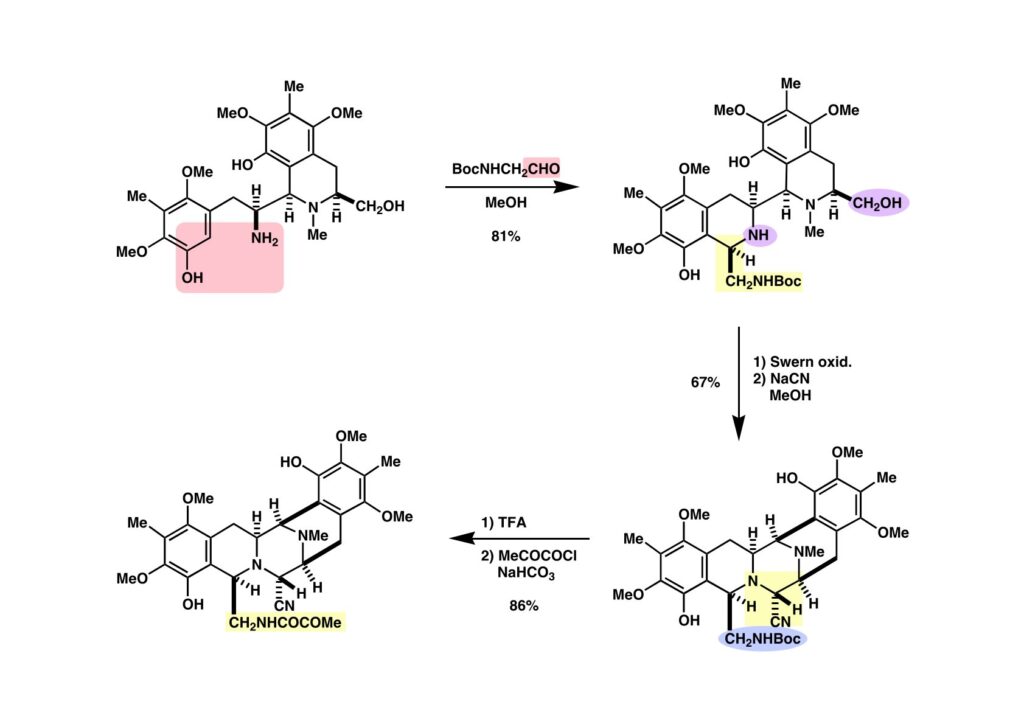 アミノフェノール (1-1) とアルデヒド (1-2) のPictet-Spengler反応は問題なく進行し、ほぼ目的とするβ-体 (1-2) のみが得られた。フェノール2つとフリーのアミンが存在するアルコール (1-2) の酸化は容易ではなかったが、幸いSwern酸化を注意深く行うことにより不安定なヘミアミナールを生成したが、NaCNを加えて、より安定なアミノニトリル (2-2) として単離することができた。次にBoc基をTFAで落として、pyruvoyl chlorideを用いて (2-1) に変換した。
アミノフェノール (1-1) とアルデヒド (1-2) のPictet-Spengler反応は問題なく進行し、ほぼ目的とするβ-体 (1-2) のみが得られた。フェノール2つとフリーのアミンが存在するアルコール (1-2) の酸化は容易ではなかったが、幸いSwern酸化を注意深く行うことにより不安定なヘミアミナールを生成したが、NaCNを加えて、より安定なアミノニトリル (2-2) として単離することができた。次にBoc基をTFAで落として、pyruvoyl chlorideを用いて (2-1) に変換した。
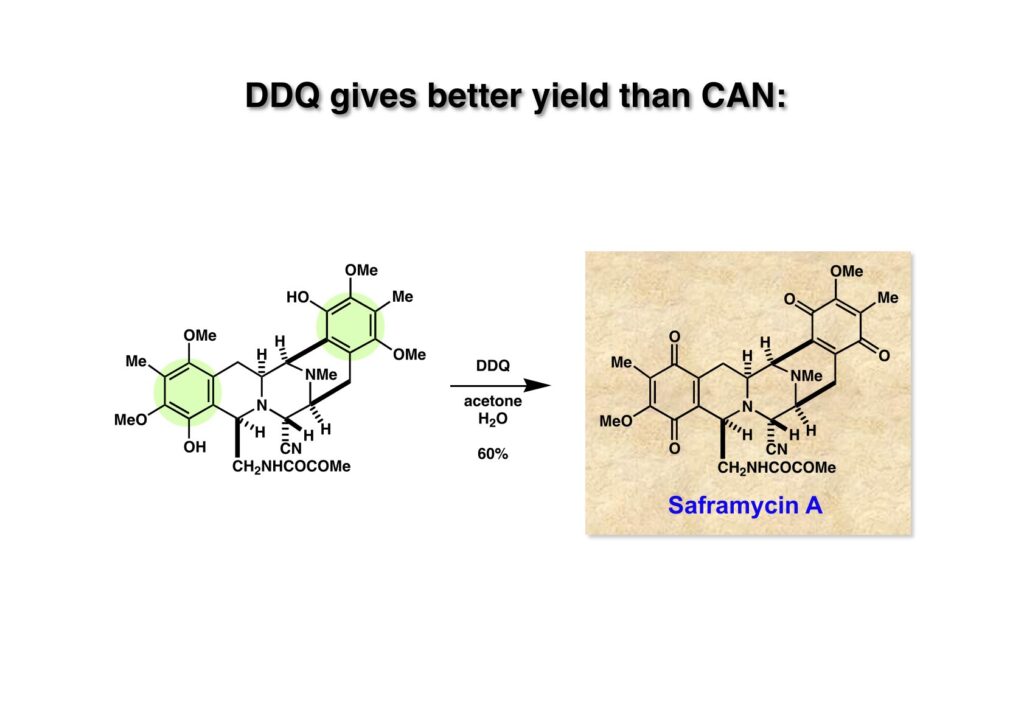 最後に残った二つのフェノール (1-1) をDDQで酸化することにより、目的とするsaframycin A (1-2) の合成に成功した。Saframycin Aは特に愛着を持った化合物ではなかったが、終わってやれやれという感じだった。ちょっとダラダラとやり過ぎたかもしれない。
最後に残った二つのフェノール (1-1) をDDQで酸化することにより、目的とするsaframycin A (1-2) の合成に成功した。Saframycin Aは特に愛着を持った化合物ではなかったが、終わってやれやれという感じだった。ちょっとダラダラとやり過ぎたかもしれない。