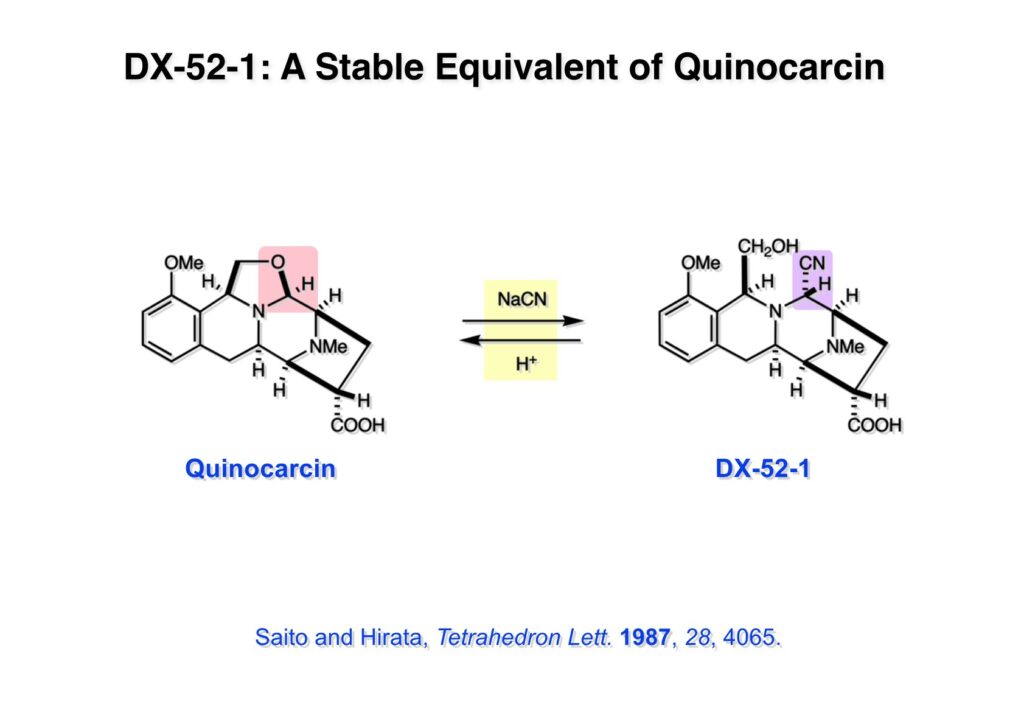
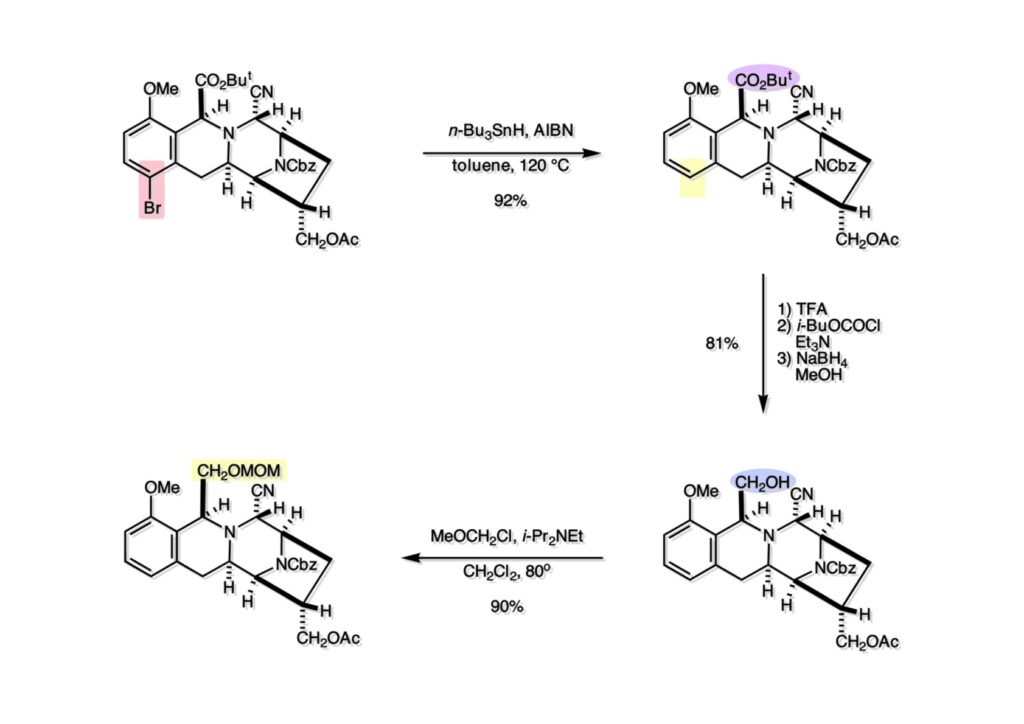
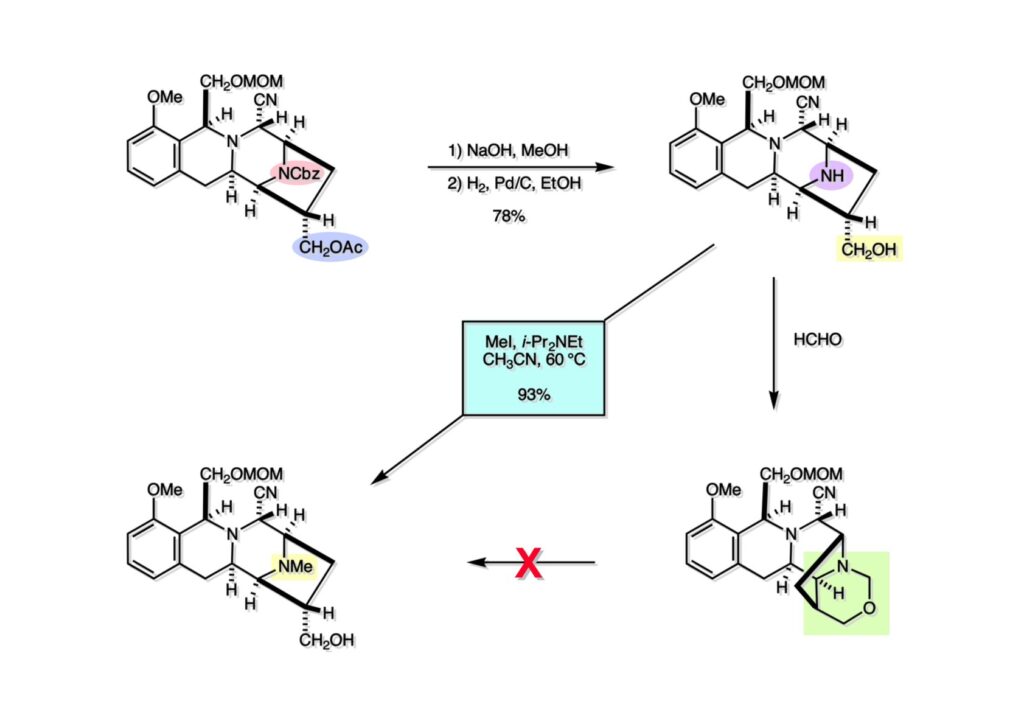
Quinocarcinの全合成はnaphthyridinomycinの全合成研究をやっていなければ着手しなかったプロジェクトである。後者のモデル実験をやっている時に、これはquinocarcinにも使えると思い合成を開始した。Joe Nunesというカーネギーメロン大学出身の学生で、物静かな真面目な若者で根性も十分備えていた。カーネギーメロン大化学科の助教授だった目さんに勧められて私の研究室に来た。目さんはWoodward研のポスドクをされている時に重なっていて、erythromycinの全合成チームに属していた。威勢の良い方だったが若くして亡くなられたのは残念である。
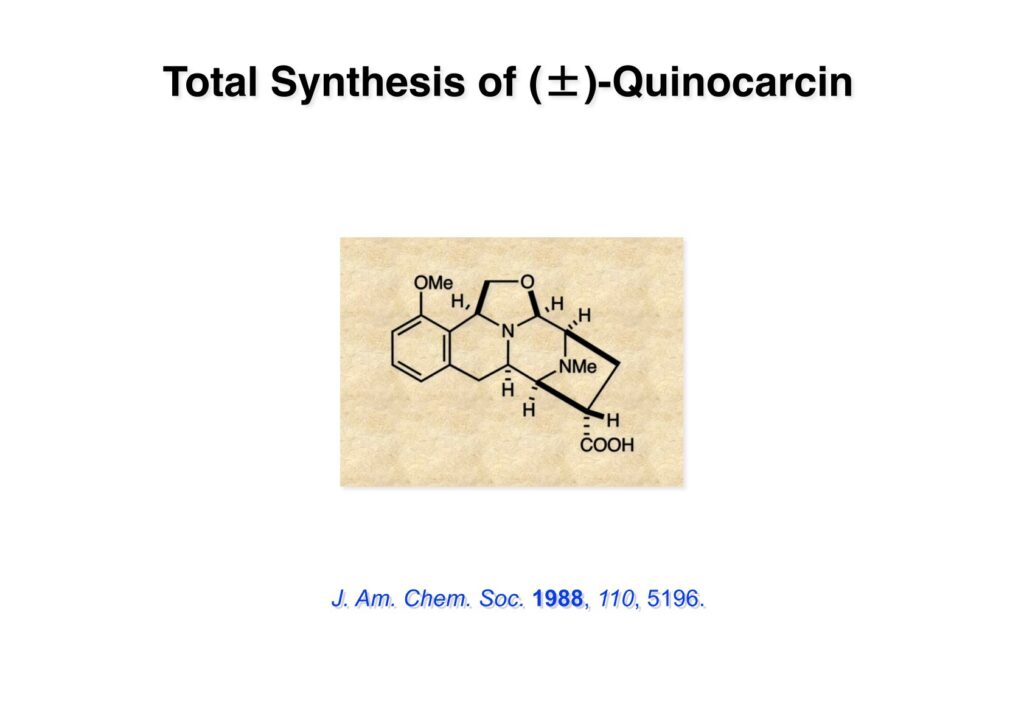
“Stereocontrolled Total Synthesis of (±)-Quinocarcin,” T. Fukuyama and J. J. Nunes, J. Am. Chem. Soc., 110, 5196 (1988).
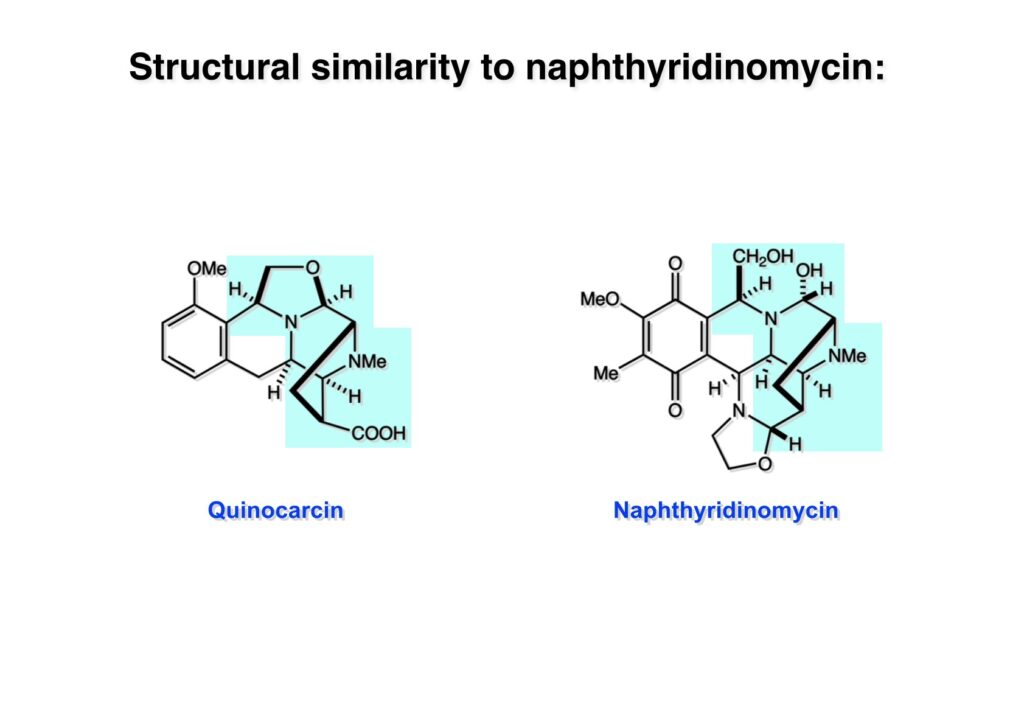
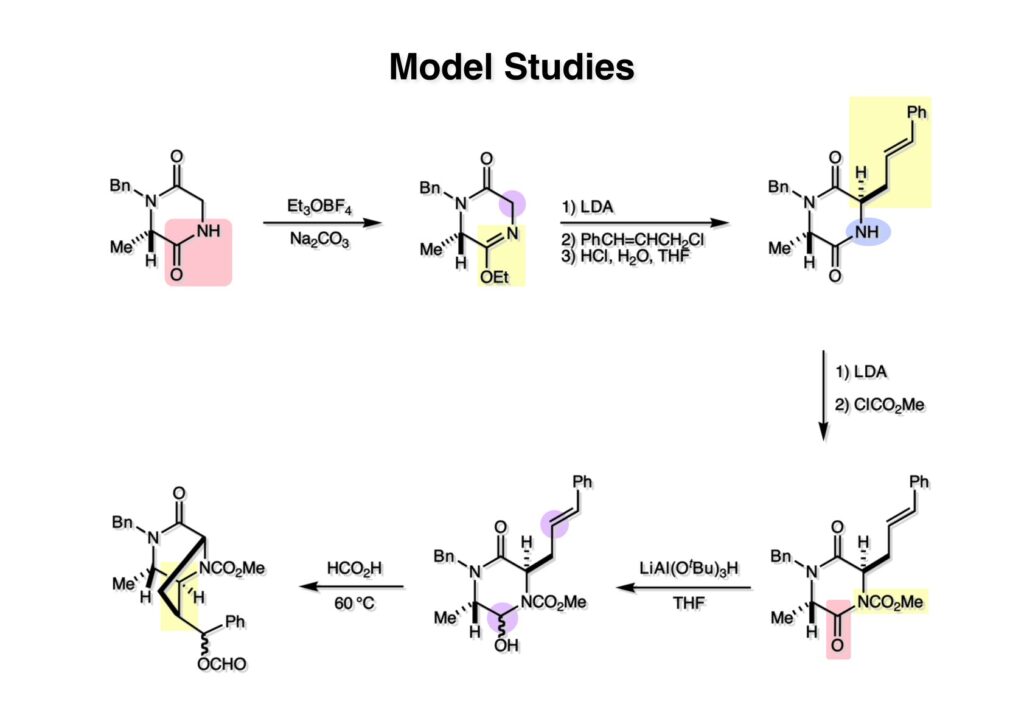
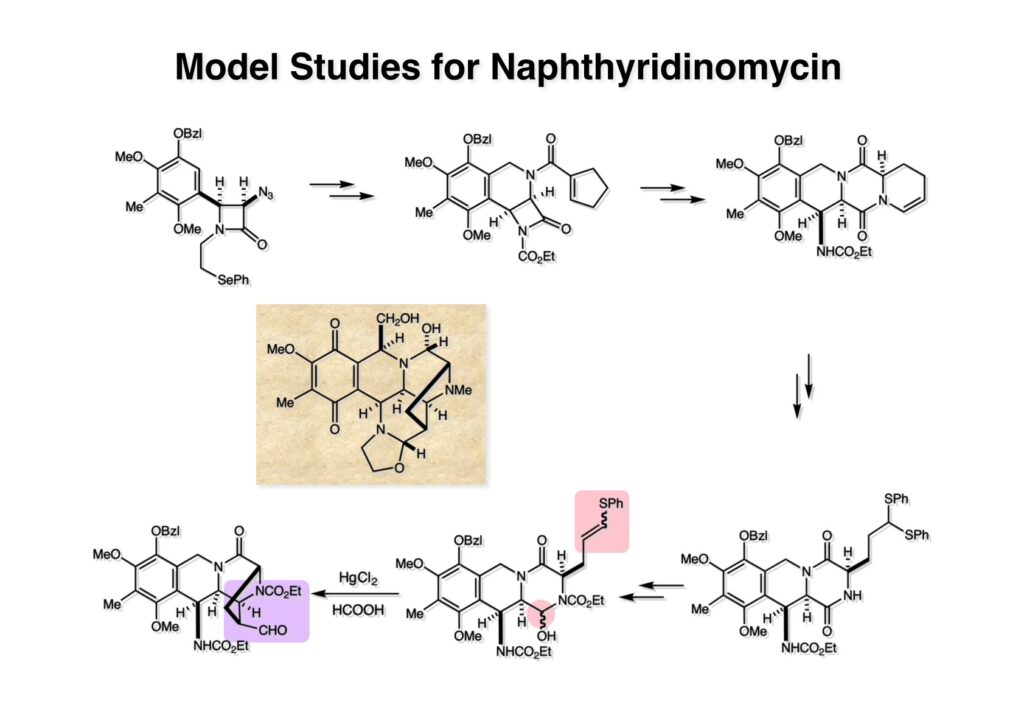
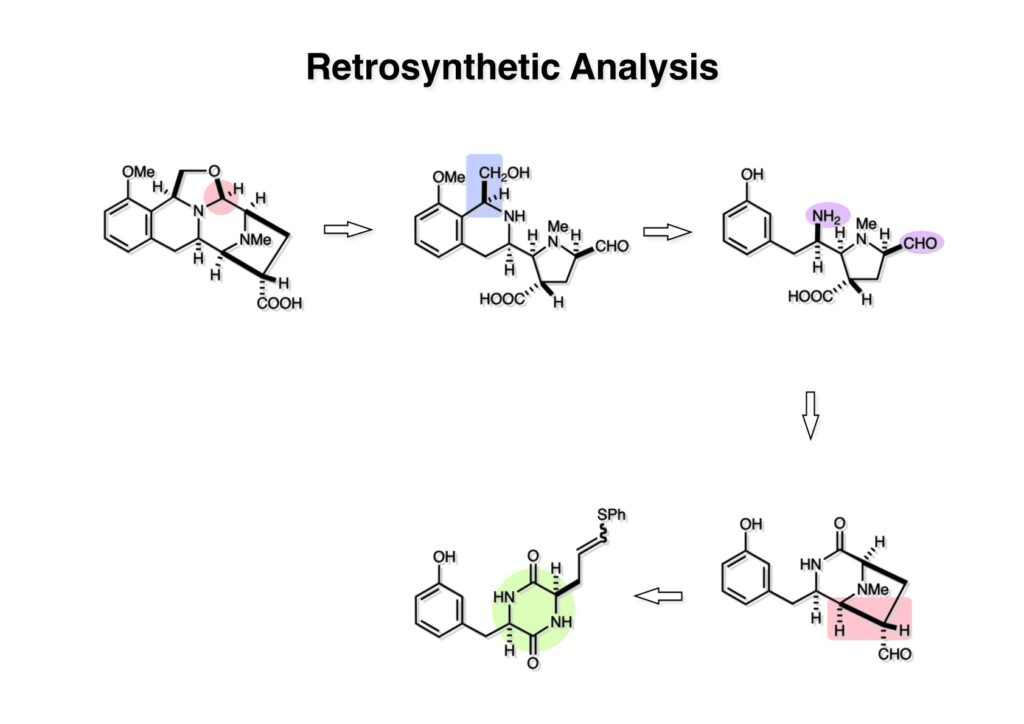


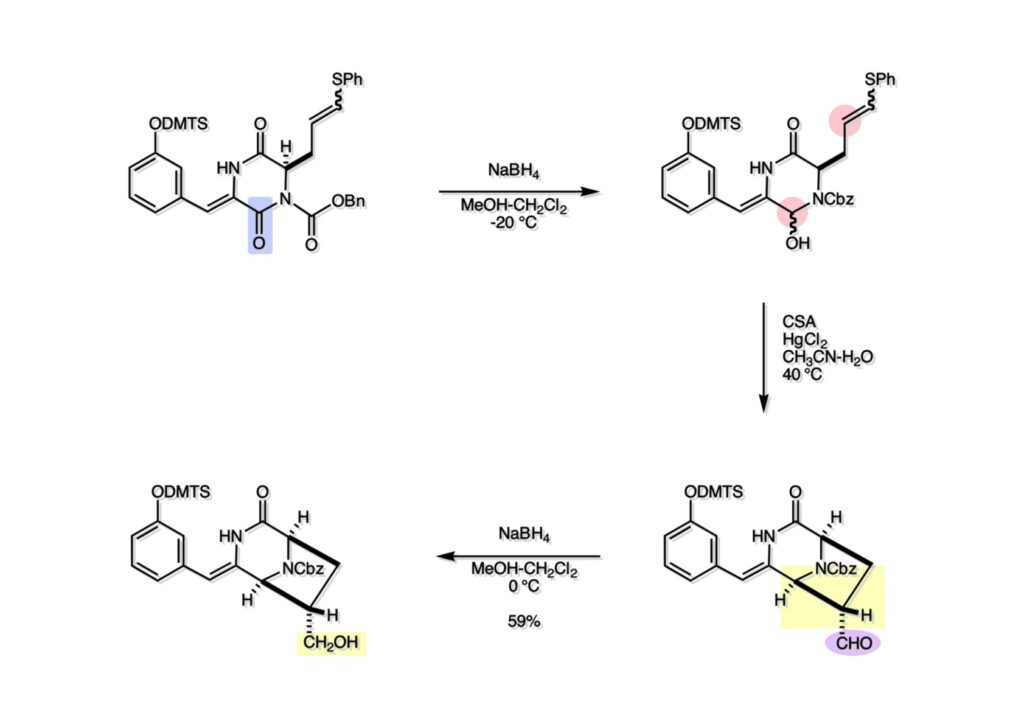
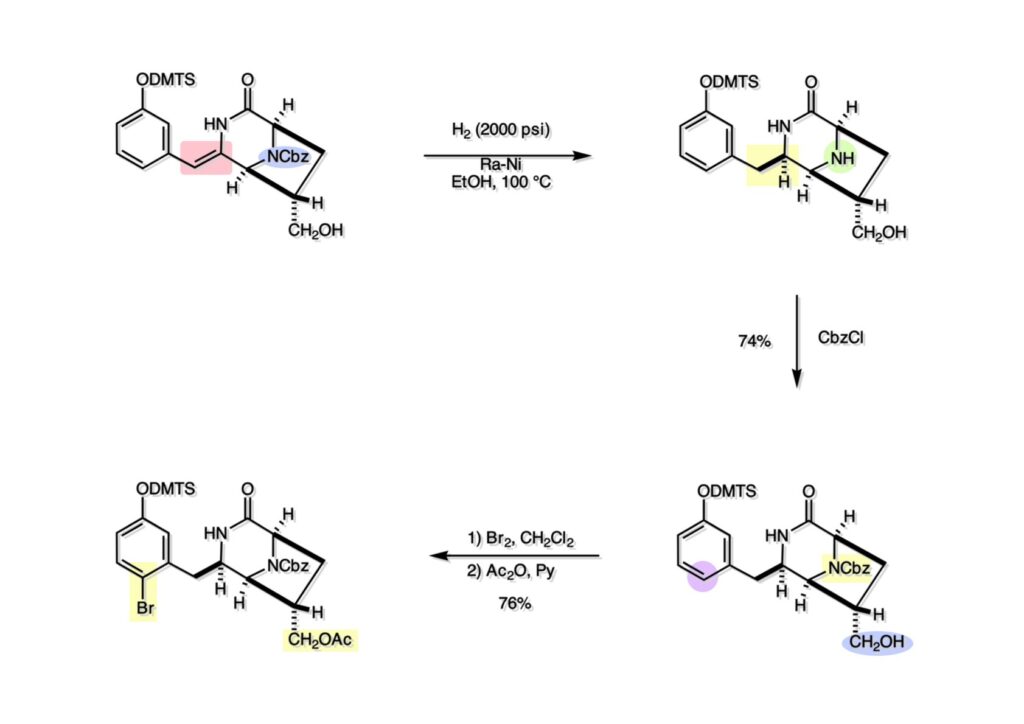
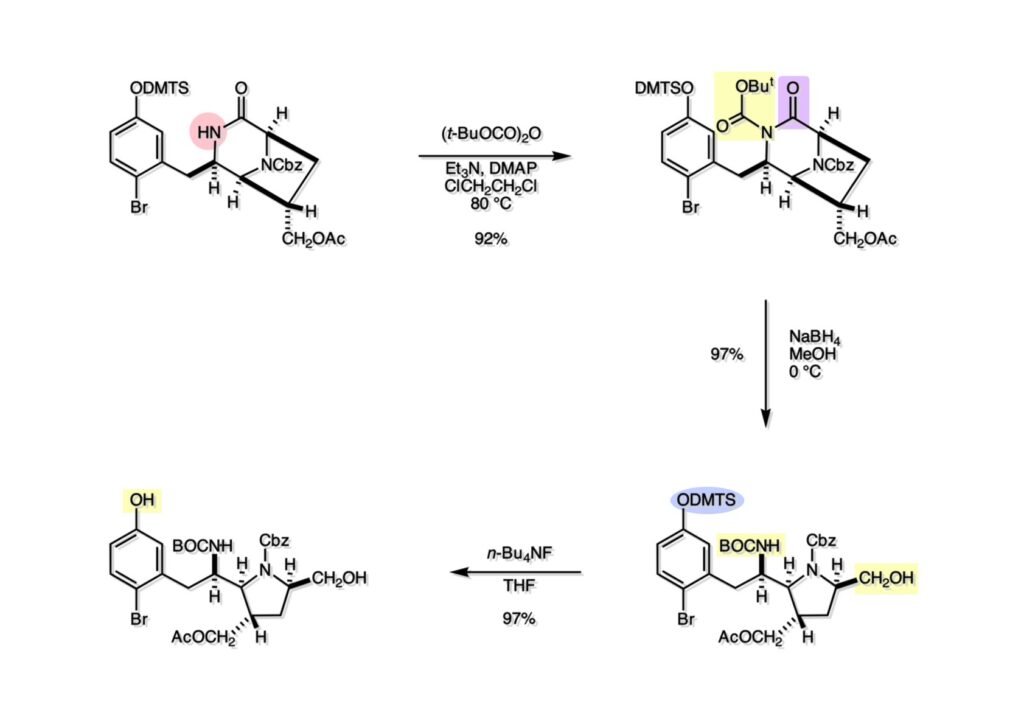
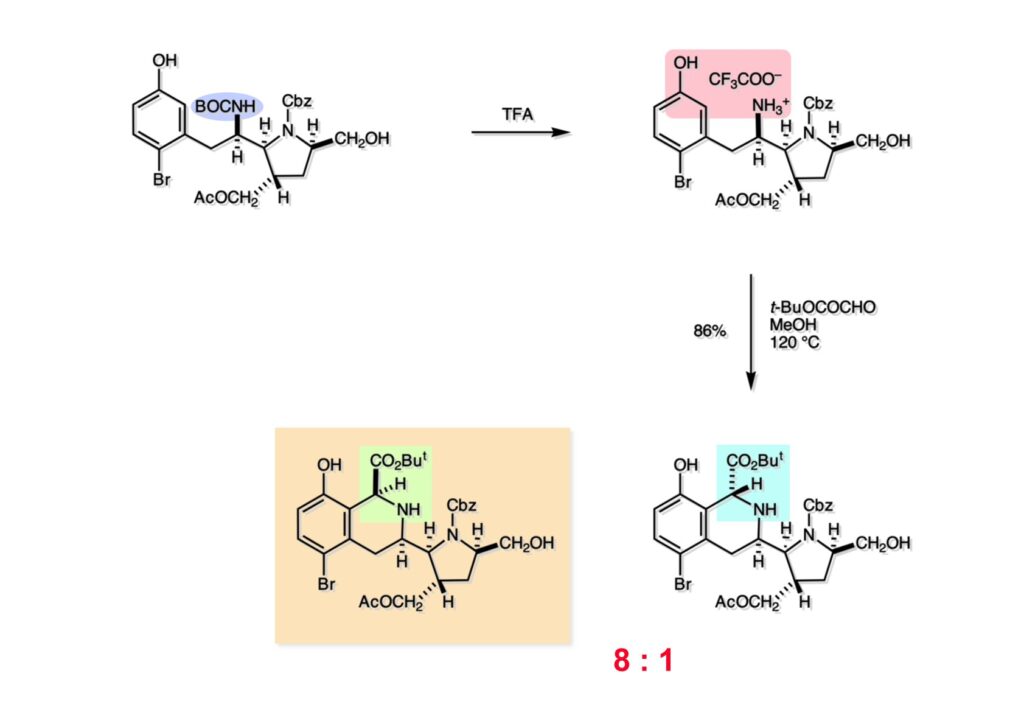
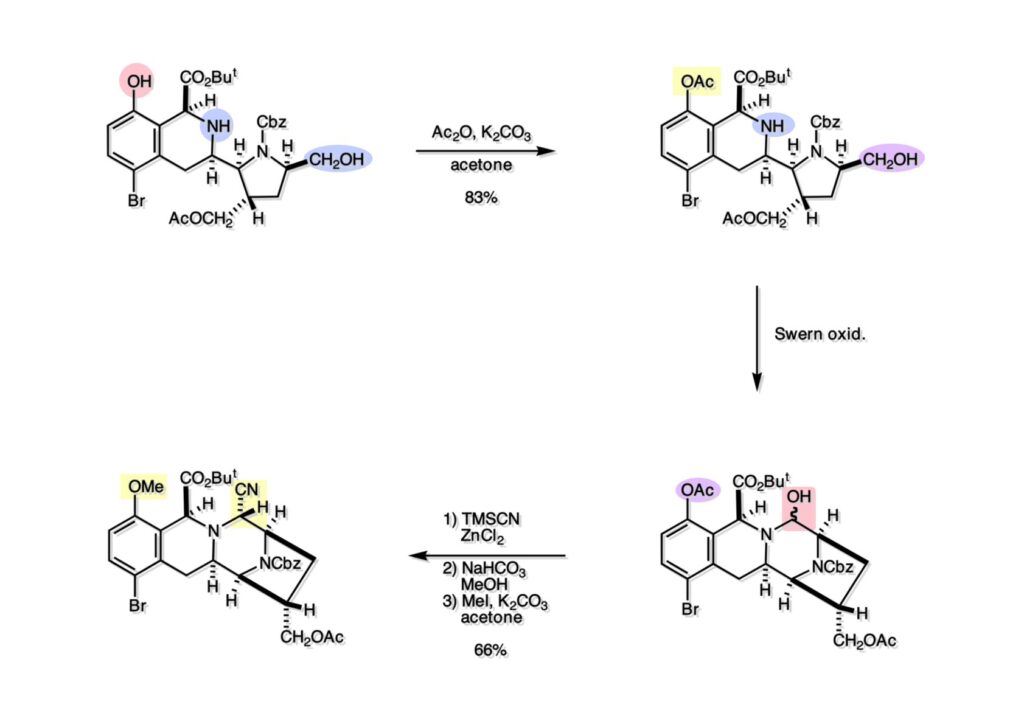
(1-1) のNaBH4還元で得たヘミアミナール誘導体 (1-2) を酸性条件に付すとアシルイミニウムイオン生成を経てビシクロ体 (2-2) を与えた。ここで通常のギ酸中加熱の条件を用いなかったのは、生成物としてアルデヒドを直接得たかったからで、HgCl2を共存させたのはPhSHをトラップしてチオアセタール生成を抑えるためであった。アルデヒドは後に酸化してカルボン酸にするのだが、ここでは扱いやすいアルコール (2-1) に還元しておいた。