Leustroducsin Bは血小板増多作用を持つ新規抗生物質として三共株式会社で放線菌から単離構造決定された化合物である。東大薬の廣部雅昭先生の研究室で修士号を取得後、三共の研究員をしていた島田神生さんが私の研究室に2年間研究生として来られることになった。経緯は忘れてしまったが島田さんがこの化合物の全合成をやりたいと言ったのだと思う。私が含窒素天然物全合成の専門家と言っても、leustroducsin Bにはアミノ基が一個あるだけなのでほぼ専門外の化合物と言って良い。この天然物は酸にもアルカリにも弱く、保護基をどのように使うかというのが課題となった。一人で2年以内に完成させるのはさすがに厳しかったので、後半は修士課程の学生だった鏑木洋介君に助太刀をしてもらい完成に漕ぎ着けた。
“Total Synthesis of Leustroducsin B,” K. Shimada, Y. Kaburagi, and T. Fukuyama, J. Am. Chem. Soc., 125, 4048 (2003).

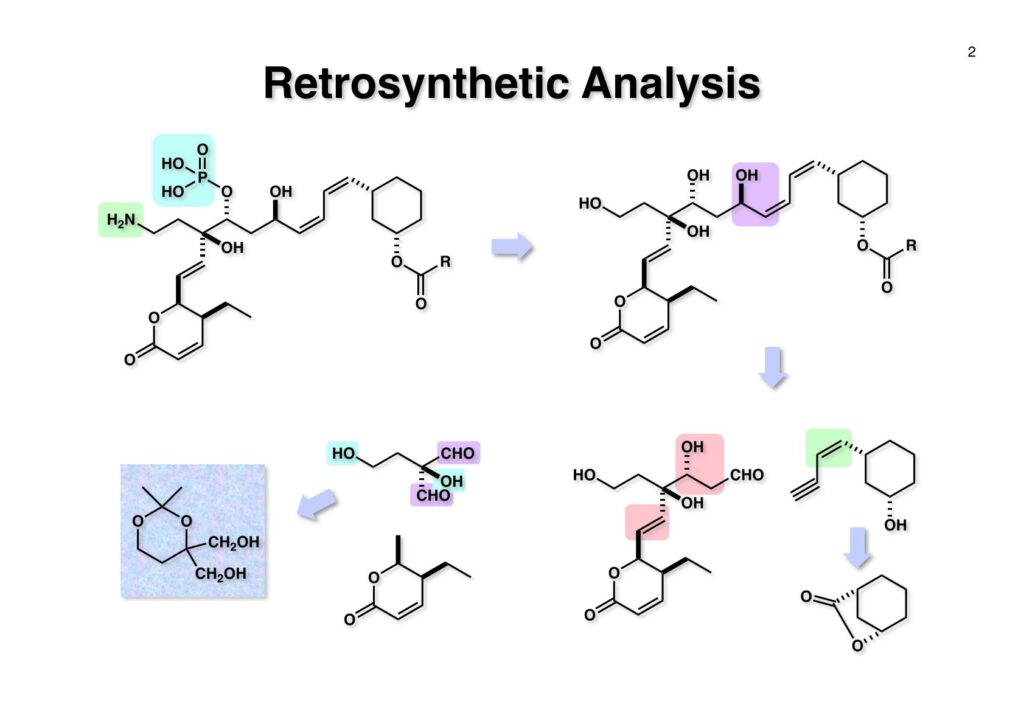 逆合成解析を簡単に示すが、まず (1-1) のアミノ基やリン酸基は合成途上で邪魔になるので、なるべく全合成の後半に導入することにしたい。次に (1-2) を眺めてみると、ジエンはcis-cisなのでアセチレンの還元で立体化学を制御できるし、アセチリドのアルデヒドへの付加ではキレーション制御で水酸基の立体化学を制御できる可能性がある。従って 、(1-2) は (2-3) と (2-4) に分割することができる。(2-4) の置換基のcis配置は (3-2) のようなラクトンから導くことを期待した。(2-3) のβ-ヒドロキシアルデヒドとジヒドロピラノン部分はいずれも (2-2) のアルデヒドから構築できると考えれば (2-1) のようなジオール体が重要な出発点となることが考えられる。この化合物は対象面と4置換炭素を有しており、不斉合成では非対称化が困難で、すぐに思い浮かんだのは酵素にお願いすることだった。
逆合成解析を簡単に示すが、まず (1-1) のアミノ基やリン酸基は合成途上で邪魔になるので、なるべく全合成の後半に導入することにしたい。次に (1-2) を眺めてみると、ジエンはcis-cisなのでアセチレンの還元で立体化学を制御できるし、アセチリドのアルデヒドへの付加ではキレーション制御で水酸基の立体化学を制御できる可能性がある。従って 、(1-2) は (2-3) と (2-4) に分割することができる。(2-4) の置換基のcis配置は (3-2) のようなラクトンから導くことを期待した。(2-3) のβ-ヒドロキシアルデヒドとジヒドロピラノン部分はいずれも (2-2) のアルデヒドから構築できると考えれば (2-1) のようなジオール体が重要な出発点となることが考えられる。この化合物は対象面と4置換炭素を有しており、不斉合成では非対称化が困難で、すぐに思い浮かんだのは酵素にお願いすることだった。
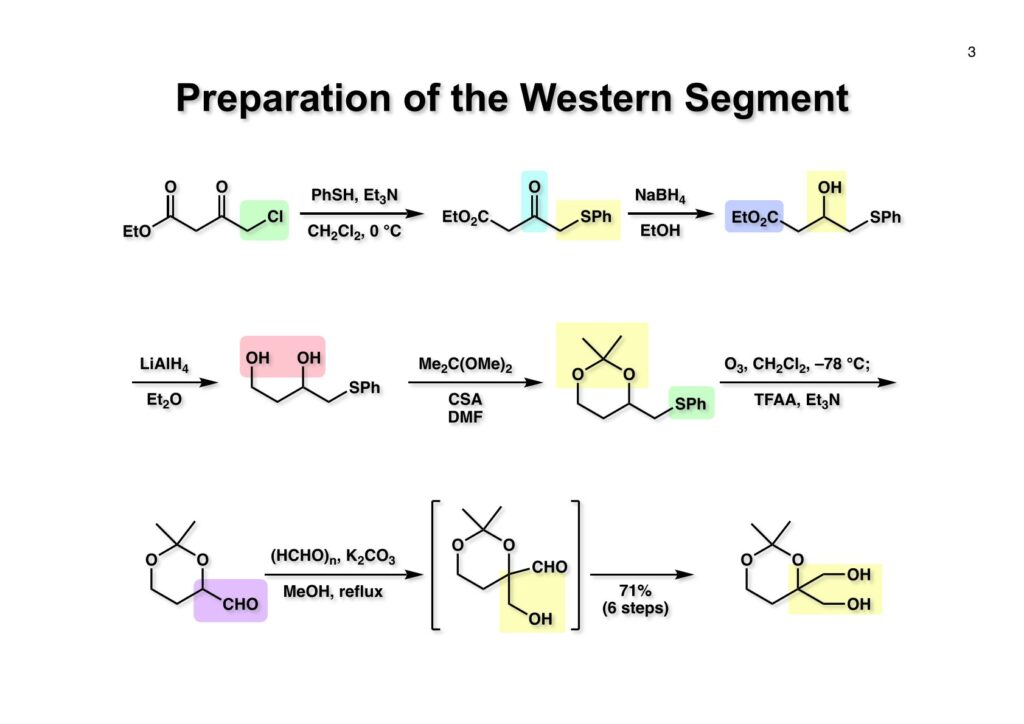 少々まどろっこしいが、左側のパート (3-3) は大量合成が容易にできるルートをデザインした。まず比較的廉価なethyl 4-chloroacetoacetate (1-1) をチオフェノールとEt3Nでサルファイド (1-2) に変換し、ケトンをNaBH4還元してアルコール (1-3) を得た。次にエステルをLAH還元してジオール (2-1) にしてからアセトナイド (2-2) で保護した。次はスルホキサイドにしてからPummerer転位をするのだが酸化にはオゾンを用いた。NaIO4では遅すぎるし、MCPBAでは注意深くやらないとスルホンが副生してしまう。オゾンを用いると低温下では付加体が生成してそれ以上反応が起きず、昇温することでスルホキサイドとおそらく一重項酸素が生成すると思われる。ハーバード大学のPaul Bartlett教授(講義を受講したことがある)は(PhO)3Pに低温下でオゾンを反応させて(PhO)3PO3にし、昇温することで一重項酸素を発生させていた。オゾン酸化はCH2Cl2を溶媒にし、後処理の必要が無いので、昇温後に無水トリフロロ酢酸とEt3Nを加えて後処理するだけでアルデヒド (3-1) が得られる。これをメタノール中で過剰のパラホルムアルデヒドとK2CO3で加熱環流すると、まずヒドロキシメチル化で (3-2) が生成し、次にCannizzaro反応でアルデヒドが還元されて目的とするビスヒドロキシメチル体 (3-3) が (1-1) から71%の通算収率で得られた。
少々まどろっこしいが、左側のパート (3-3) は大量合成が容易にできるルートをデザインした。まず比較的廉価なethyl 4-chloroacetoacetate (1-1) をチオフェノールとEt3Nでサルファイド (1-2) に変換し、ケトンをNaBH4還元してアルコール (1-3) を得た。次にエステルをLAH還元してジオール (2-1) にしてからアセトナイド (2-2) で保護した。次はスルホキサイドにしてからPummerer転位をするのだが酸化にはオゾンを用いた。NaIO4では遅すぎるし、MCPBAでは注意深くやらないとスルホンが副生してしまう。オゾンを用いると低温下では付加体が生成してそれ以上反応が起きず、昇温することでスルホキサイドとおそらく一重項酸素が生成すると思われる。ハーバード大学のPaul Bartlett教授(講義を受講したことがある)は(PhO)3Pに低温下でオゾンを反応させて(PhO)3PO3にし、昇温することで一重項酸素を発生させていた。オゾン酸化はCH2Cl2を溶媒にし、後処理の必要が無いので、昇温後に無水トリフロロ酢酸とEt3Nを加えて後処理するだけでアルデヒド (3-1) が得られる。これをメタノール中で過剰のパラホルムアルデヒドとK2CO3で加熱環流すると、まずヒドロキシメチル化で (3-2) が生成し、次にCannizzaro反応でアルデヒドが還元されて目的とするビスヒドロキシメチル体 (3-3) が (1-1) から71%の通算収率で得られた。
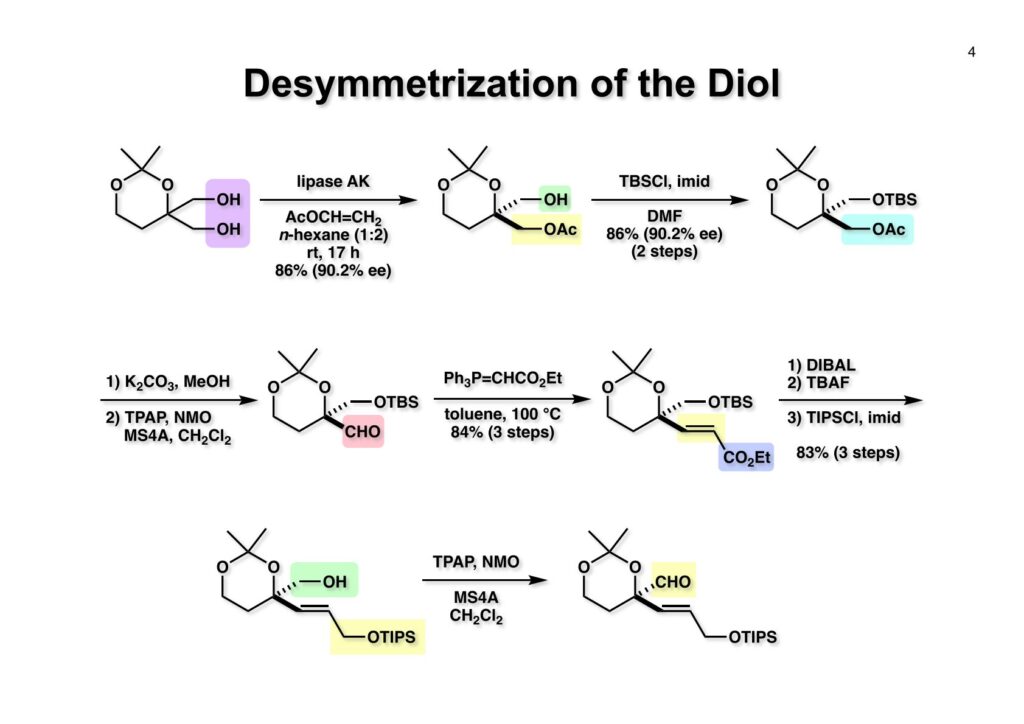 ジオール (1-1) の非対称化は最初はジアセテートのリパーゼによる選択的加水分解を試したが良い結果は得られなかった。そこで酢酸ビニルを用いてジオールのアセチル化を行ったところ、lipase AKで酢酸ビニル:n-ヘキサン(1:2)中、室温で17時間攪拌することで (1-2) が86% (90.2% ee) の収率で得られた。当研究室で用いた酵素は殆どが天野エンザイムの広瀬芳彦氏から恵与されたものである。(1-2) のアセテートは放置しておくとアセチル基がキャッチボールされてラセミ化が進行するので、反応終了後は直ちに水酸基をTBS化して (1-3) を得た。(1-3) のアセテートは加メタノール分解し、得られたヒドロキシメチル基をSteve LeyのTPAP酸化の条件に付してアルデヒド (2-1) に変換した。先ほどアセチル基が転移しやすいことをコメントしたが、実はシリル基も条件によっては同様に転移するので油断はならない。アルデヒド (2-1) を安定イリドによるWittig反応で不飽和エステル (2-2) にし、DIBAL還元とTBAFによる脱TBS化でジオールに変換し、さらに嵩高いTIPSClで立体的に空いているアリルアルコールを選択的に保護して (3-1) を得た。ヒドロキシメチル体 (3-1) を再びTPAP酸化することでアルデヒド (3-2) が得られた。
ジオール (1-1) の非対称化は最初はジアセテートのリパーゼによる選択的加水分解を試したが良い結果は得られなかった。そこで酢酸ビニルを用いてジオールのアセチル化を行ったところ、lipase AKで酢酸ビニル:n-ヘキサン(1:2)中、室温で17時間攪拌することで (1-2) が86% (90.2% ee) の収率で得られた。当研究室で用いた酵素は殆どが天野エンザイムの広瀬芳彦氏から恵与されたものである。(1-2) のアセテートは放置しておくとアセチル基がキャッチボールされてラセミ化が進行するので、反応終了後は直ちに水酸基をTBS化して (1-3) を得た。(1-3) のアセテートは加メタノール分解し、得られたヒドロキシメチル基をSteve LeyのTPAP酸化の条件に付してアルデヒド (2-1) に変換した。先ほどアセチル基が転移しやすいことをコメントしたが、実はシリル基も条件によっては同様に転移するので油断はならない。アルデヒド (2-1) を安定イリドによるWittig反応で不飽和エステル (2-2) にし、DIBAL還元とTBAFによる脱TBS化でジオールに変換し、さらに嵩高いTIPSClで立体的に空いているアリルアルコールを選択的に保護して (3-1) を得た。ヒドロキシメチル体 (3-1) を再びTPAP酸化することでアルデヒド (3-2) が得られた。
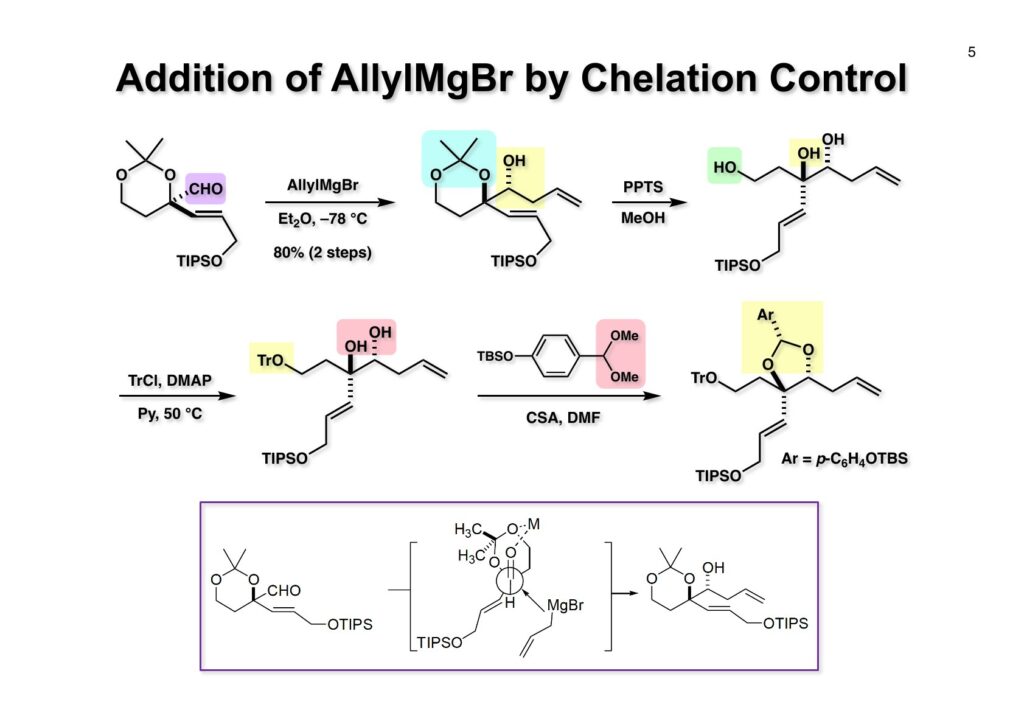 アルデヒドやケトンのα位やβ位に酸素原子など金属イオンに配位できる官能基が存在するとキレーションによって立体化学を制御することができる。(1-1) のエーテル溶液を–78 °Cに冷却してallylMgBrを滴下すると (1-2) が>95:<5の比で得られた。下段に島田さんの博士論文からパクった立体制御の図を挿入しておいた。アセトナイド (1-2) の除去はメタノール中でPPTSを使って行いトリオール (1-3) を得た。1級アルコールの保護は定番のトリチル基を用いた。(2-1) のジオールの保護は通常のアセトナイドやアルデヒドのアセタールを用いても合成後期の肝心なステージで選択的に脱保護することが出来なかったので自前の保護基を考えることにした。その結果が (2-2) のp-TBSOC6H4CH(OMe)2である。フェノールのTBSエーテルは強塩基には弱いが酸性条件ではかなり安定である。おそらくフェノールの酸素原子はプロトネーションが起きにくいからだろう。ジオール (2-1) はアセタール (2-2) とCSAで50 °Cに加熱することでアセタール (2-3) が得られた。(2-3) のアリール基の立体化学はNOEで決定した。
アルデヒドやケトンのα位やβ位に酸素原子など金属イオンに配位できる官能基が存在するとキレーションによって立体化学を制御することができる。(1-1) のエーテル溶液を–78 °Cに冷却してallylMgBrを滴下すると (1-2) が>95:<5の比で得られた。下段に島田さんの博士論文からパクった立体制御の図を挿入しておいた。アセトナイド (1-2) の除去はメタノール中でPPTSを使って行いトリオール (1-3) を得た。1級アルコールの保護は定番のトリチル基を用いた。(2-1) のジオールの保護は通常のアセトナイドやアルデヒドのアセタールを用いても合成後期の肝心なステージで選択的に脱保護することが出来なかったので自前の保護基を考えることにした。その結果が (2-2) のp-TBSOC6H4CH(OMe)2である。フェノールのTBSエーテルは強塩基には弱いが酸性条件ではかなり安定である。おそらくフェノールの酸素原子はプロトネーションが起きにくいからだろう。ジオール (2-1) はアセタール (2-2) とCSAで50 °Cに加熱することでアセタール (2-3) が得られた。(2-3) のアリール基の立体化学はNOEで決定した。
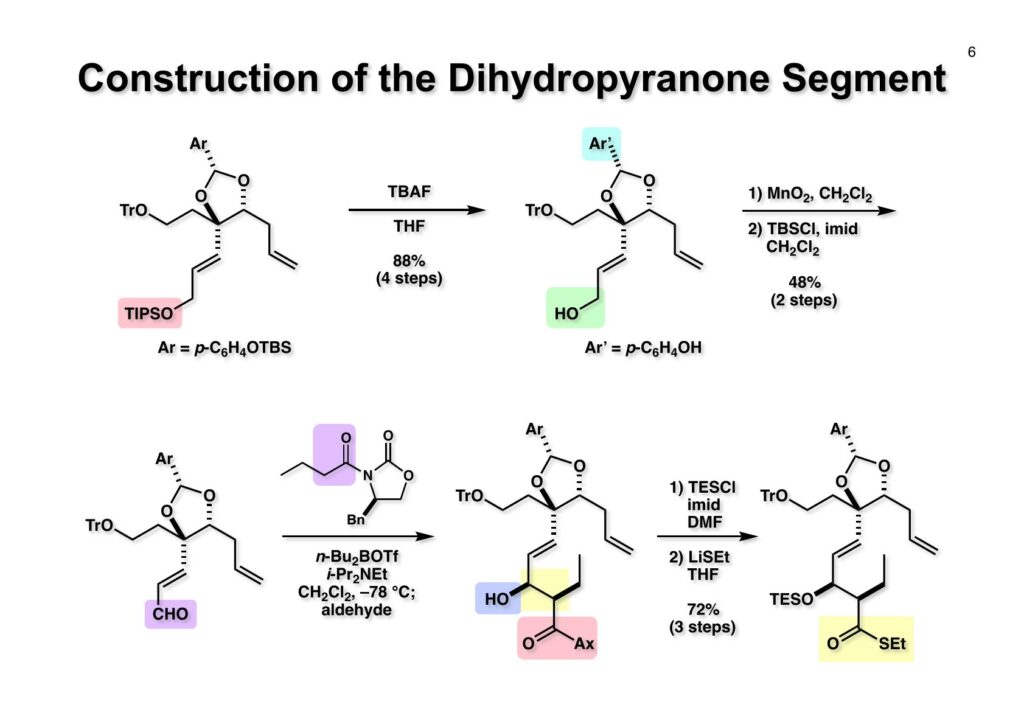 次はジヒドロピラノン部分の構築に取りかかった。環上に二つのキラル中心があるのでEvansの不斉補助基を用いたアルドール型の反応を利用することにした。まず (1-1) のTIPS基をTBAFで除去し、アリルアルコーの選択的酸化に常用されるMnO2を用いてアルデヒドに変換した。フェノール上のTBS基が外れてしまっているので保護し直して (2-1) を得た。ここで (2-2) を用いてEvansのジアステレオ選択的なアルドール型反応を行って付加体 (2-3) を得た。2ページ前の酵素を使ったジオールの非対称化で生じたエナンチオマーに起因するジアステレオマーはこの段階で分離し、(2-3) は光学的に純粋な化合物となった。(2-3) の水酸基をTES基で保護した後にLiSEtを用いて不斉補助基を除去してチオエステル (2-4) を得た。
次はジヒドロピラノン部分の構築に取りかかった。環上に二つのキラル中心があるのでEvansの不斉補助基を用いたアルドール型の反応を利用することにした。まず (1-1) のTIPS基をTBAFで除去し、アリルアルコーの選択的酸化に常用されるMnO2を用いてアルデヒドに変換した。フェノール上のTBS基が外れてしまっているので保護し直して (2-1) を得た。ここで (2-2) を用いてEvansのジアステレオ選択的なアルドール型反応を行って付加体 (2-3) を得た。2ページ前の酵素を使ったジオールの非対称化で生じたエナンチオマーに起因するジアステレオマーはこの段階で分離し、(2-3) は光学的に純粋な化合物となった。(2-3) の水酸基をTES基で保護した後にLiSEtを用いて不斉補助基を除去してチオエステル (2-4) を得た。
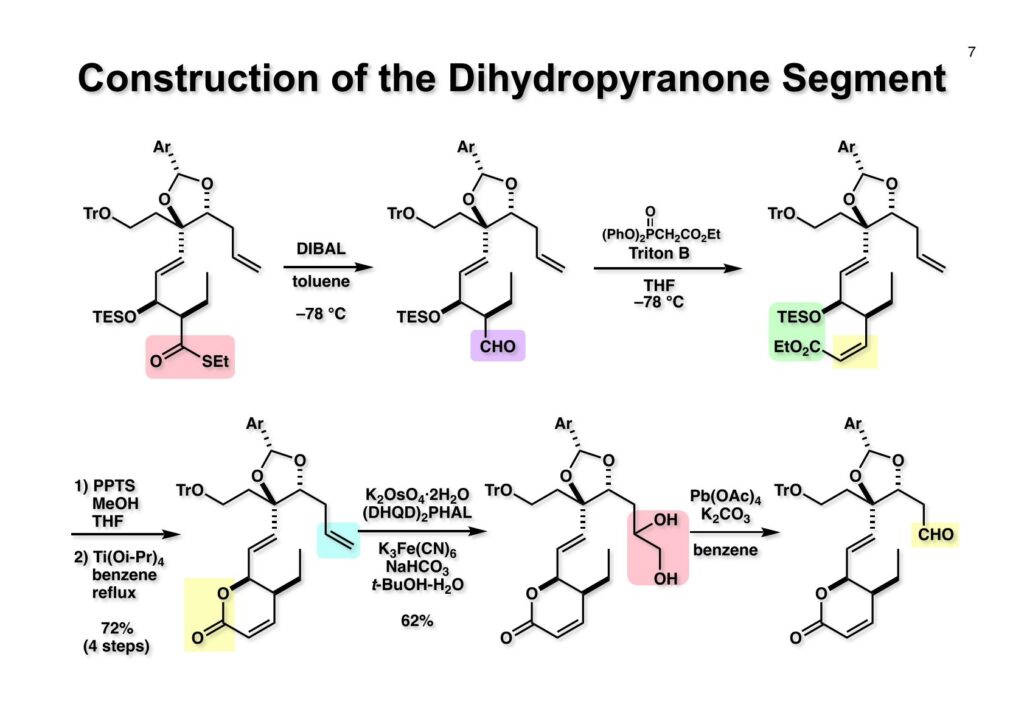 チオエステル (1-1) のアルデヒドへの還元はEt3SiH-Pd/Cでやろうと思ったが末端オレフィンが還元されてしまったのでDIBALを低温下で用いて (1-2) を得た。還元されやすいオレフィンが存在している場合の福山還元にはLindlar触媒を用いれば良いとEvansが報告しているが、他にDIBALで還元されるところが無いのでそれを使った次第。このアルデヒドからcis-α,β-不飽和エステルに変換するのに当時琉球大学に在籍していた安藤香織さんが報告した試薬を活用した。得られた (1-3) のTES基をMeOH-THF中弱酸のPPTSを使うことで除去し、次にTi(Oi-Pr)4を用いてベンゼン中加熱環流することでラクトン (2-1) に変換した。トリチル基やアセタール存在下では強い酸を使ってラクトン化することが出来ないため、中性に近い条件でアルコールを活性化した。3つのオレフィンが混在する (2-1) の末端オレフィンのジヒドロキシ化は、不斉反応の必要は無いが選択性の良いSharplessのAD-mix-βを用いた。得られたジオール (2-2) は四酢酸鉛で開裂させてアルデヒド (2-3) を得た。
チオエステル (1-1) のアルデヒドへの還元はEt3SiH-Pd/Cでやろうと思ったが末端オレフィンが還元されてしまったのでDIBALを低温下で用いて (1-2) を得た。還元されやすいオレフィンが存在している場合の福山還元にはLindlar触媒を用いれば良いとEvansが報告しているが、他にDIBALで還元されるところが無いのでそれを使った次第。このアルデヒドからcis-α,β-不飽和エステルに変換するのに当時琉球大学に在籍していた安藤香織さんが報告した試薬を活用した。得られた (1-3) のTES基をMeOH-THF中弱酸のPPTSを使うことで除去し、次にTi(Oi-Pr)4を用いてベンゼン中加熱環流することでラクトン (2-1) に変換した。トリチル基やアセタール存在下では強い酸を使ってラクトン化することが出来ないため、中性に近い条件でアルコールを活性化した。3つのオレフィンが混在する (2-1) の末端オレフィンのジヒドロキシ化は、不斉反応の必要は無いが選択性の良いSharplessのAD-mix-βを用いた。得られたジオール (2-2) は四酢酸鉛で開裂させてアルデヒド (2-3) を得た。
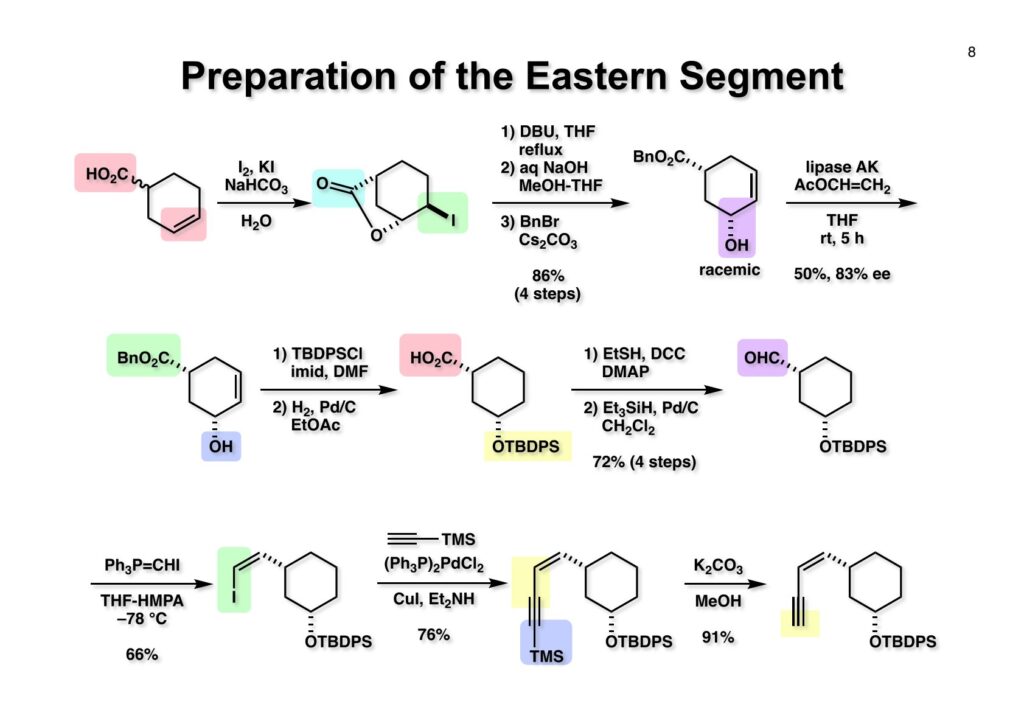 右側の部分も光学活性体を作らなければならないが、アクリル酸誘導体とブタジエンのDiels-Alder反応でラセミ体は簡単に作れるものの不斉合成で実用的なものは無かったと思う。今では (1-1) の光学活性体が50g·3万円で市販されているので酵素を使う必要はないが、まあ20年以上前のことだから仕方ない。ラセミ体の (1-1) をiodolactonizationすると簡単に (1-2) が得られる。これをDBUを用いて脱HI化し、ラクトンのアルカリ加水分解、カルボン酸のベンジルエステル化を経て (1-3) を得た。Lipase AKによる (1-3) の光学分割は小スケールでは40% (96% ee) の結果が得られたものの77 gスケールでは50% (83% ee) と光学純度は下がったものの、そのまま前に進むことにした。(2-1) の水酸基をTBS化した後に接触還元によってカルボン酸 (2-2) を得た。このカルボン酸をチオエステルに変換後に福山還元に付してアルデヒド (2-3) を得た。続くPh3P=CHIによるWittig反応はE:Z=<1:>20で望むcis体 (3-1) を与えた。TMSアセチレンとヨウ化ビニル体との薗頭カップリングで得られた (3-2) のTMS基をメタノール中炭酸カリウムで処理することにより除去して目的とするエンイン体 (3-3) の合成を達成した。
右側の部分も光学活性体を作らなければならないが、アクリル酸誘導体とブタジエンのDiels-Alder反応でラセミ体は簡単に作れるものの不斉合成で実用的なものは無かったと思う。今では (1-1) の光学活性体が50g·3万円で市販されているので酵素を使う必要はないが、まあ20年以上前のことだから仕方ない。ラセミ体の (1-1) をiodolactonizationすると簡単に (1-2) が得られる。これをDBUを用いて脱HI化し、ラクトンのアルカリ加水分解、カルボン酸のベンジルエステル化を経て (1-3) を得た。Lipase AKによる (1-3) の光学分割は小スケールでは40% (96% ee) の結果が得られたものの77 gスケールでは50% (83% ee) と光学純度は下がったものの、そのまま前に進むことにした。(2-1) の水酸基をTBS化した後に接触還元によってカルボン酸 (2-2) を得た。このカルボン酸をチオエステルに変換後に福山還元に付してアルデヒド (2-3) を得た。続くPh3P=CHIによるWittig反応はE:Z=<1:>20で望むcis体 (3-1) を与えた。TMSアセチレンとヨウ化ビニル体との薗頭カップリングで得られた (3-2) のTMS基をメタノール中炭酸カリウムで処理することにより除去して目的とするエンイン体 (3-3) の合成を達成した。
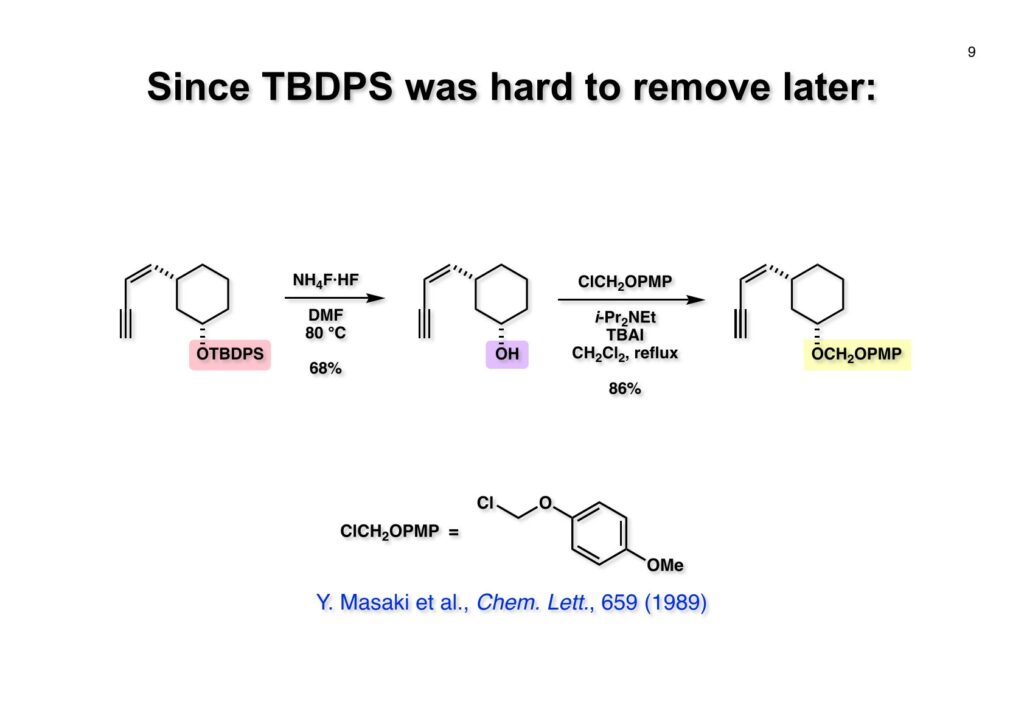 全合成終盤になってからTBDPS基の脱保護が困難であることが判明したので、酸性条件で安定なPMPOCH2基に付け替えることにした。この保護基は岐阜薬科大学の正木幸雄先生が報告したもので、PMPO基がCANによって簡単に脱保護できることは私たちが以前に報告していたが、光延反応によって導入していたので1級アルコールの保護にしか使えなかった(プロカイラルな2級アルコールは立体化学が反転してしまうので)。それをPMPOCH2基に転用するという発想が私には無かった。もちろんPMPO基の強酸に対する安定性は比類がないが。(1-1) のTBDPS基を脱保護し、得られたアルコール (1-2) を報告された条件を用いてPMPOCH2エーテル (1-3) に変換した。
全合成終盤になってからTBDPS基の脱保護が困難であることが判明したので、酸性条件で安定なPMPOCH2基に付け替えることにした。この保護基は岐阜薬科大学の正木幸雄先生が報告したもので、PMPO基がCANによって簡単に脱保護できることは私たちが以前に報告していたが、光延反応によって導入していたので1級アルコールの保護にしか使えなかった(プロカイラルな2級アルコールは立体化学が反転してしまうので)。それをPMPOCH2基に転用するという発想が私には無かった。もちろんPMPO基の強酸に対する安定性は比類がないが。(1-1) のTBDPS基を脱保護し、得られたアルコール (1-2) を報告された条件を用いてPMPOCH2エーテル (1-3) に変換した。
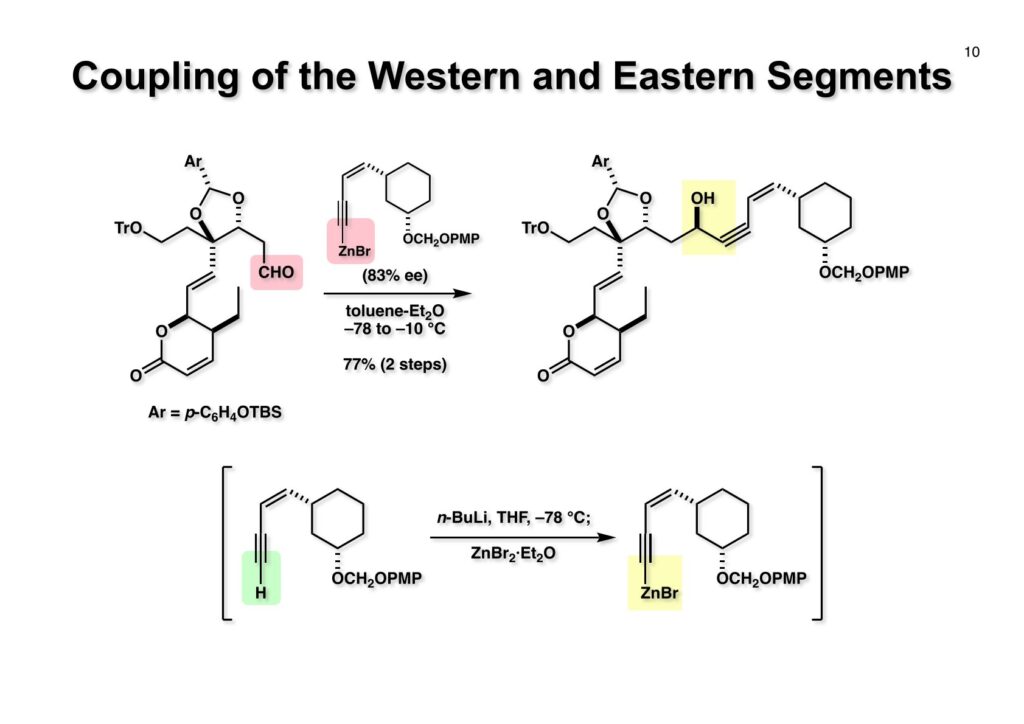 前ページで合成した (2-1) をn-BuLiで脱プロトン化し、ZnBr2·Et2Oを加えて亜鉛アセチリド (2-2) を調製した。これを–78 °Cに冷却してから (1-1) のトルエン溶液を滴下し、徐々に–10 °Cに昇温してから後処理したところ完全に立体制御された目的物 (1-2) が得られた。亜鉛によるアルデヒドとβ位のアセタールの酸素原子へのキレーションによって期待どおりの立体化学となった。不完全な酵素分割で存在していた (2-1) のエナンチオマーに起因するジアステレオマーはこの段階で分離除去された。
前ページで合成した (2-1) をn-BuLiで脱プロトン化し、ZnBr2·Et2Oを加えて亜鉛アセチリド (2-2) を調製した。これを–78 °Cに冷却してから (1-1) のトルエン溶液を滴下し、徐々に–10 °Cに昇温してから後処理したところ完全に立体制御された目的物 (1-2) が得られた。亜鉛によるアルデヒドとβ位のアセタールの酸素原子へのキレーションによって期待どおりの立体化学となった。不完全な酵素分割で存在していた (2-1) のエナンチオマーに起因するジアステレオマーはこの段階で分離除去された。
 一般的に二重結合や芳香環に結合した三重結合の部分還元は非常にトリッキーで、過還元が起きやすい。(1-1) の場合もLindlar触媒やジイミド還元でも過還元による収率の低下を招いた。第一世代のインドール合成法を開発していた時にo-iodoformanilideとプロパルギルアルコール誘導体を薗頭カップリングし、この三重結合をcis-オレフィンに還元するのにLindlar触媒では良い結果が得られなかった。ところがBrandsmaによるZn, BrCH2CH2Br, LiCuBr2/EtOH, relluxという条件を使ったところ目が覚めるように目的物だけが得られた(Aerssens, M. H. P. J.; van der Heiden, R.; Heus, M.; Brandsma, L. Synth. Commun. 1990, 20, 3421)。この切れ味の良い有用な反応がSynthetic Communicationsという、比較的マイナーな(失礼)ジャーナルに掲載されているのが信じられない。とにかく、その情報は持っていたので、この場合でも使えるかな?と思ってやってみたところ、ほぼ定量的に目的物であるジエン (1-2) が得られた。(1-2) の水酸基を保護するために種々の保護基を試したが、最終段階に近いところで満足できる収率で脱保護できるものがなかなか見つからなかった。そこでほぼ中性条件で脱保護できる可能性があるフェノキシアセテートを使うことにして (2-1) に導いた。次にアミノ基を導入するためトリチル基を外すことにした。(2-1) をZnBr2-Et3SiHで処理し、一部水酸基がTES化されてしまったのでPPTS/MeOH-THFで脱シリル化して (2-2) を得た。
一般的に二重結合や芳香環に結合した三重結合の部分還元は非常にトリッキーで、過還元が起きやすい。(1-1) の場合もLindlar触媒やジイミド還元でも過還元による収率の低下を招いた。第一世代のインドール合成法を開発していた時にo-iodoformanilideとプロパルギルアルコール誘導体を薗頭カップリングし、この三重結合をcis-オレフィンに還元するのにLindlar触媒では良い結果が得られなかった。ところがBrandsmaによるZn, BrCH2CH2Br, LiCuBr2/EtOH, relluxという条件を使ったところ目が覚めるように目的物だけが得られた(Aerssens, M. H. P. J.; van der Heiden, R.; Heus, M.; Brandsma, L. Synth. Commun. 1990, 20, 3421)。この切れ味の良い有用な反応がSynthetic Communicationsという、比較的マイナーな(失礼)ジャーナルに掲載されているのが信じられない。とにかく、その情報は持っていたので、この場合でも使えるかな?と思ってやってみたところ、ほぼ定量的に目的物であるジエン (1-2) が得られた。(1-2) の水酸基を保護するために種々の保護基を試したが、最終段階に近いところで満足できる収率で脱保護できるものがなかなか見つからなかった。そこでほぼ中性条件で脱保護できる可能性があるフェノキシアセテートを使うことにして (2-1) に導いた。次にアミノ基を導入するためトリチル基を外すことにした。(2-1) をZnBr2-Et3SiHで処理し、一部水酸基がTES化されてしまったのでPPTS/MeOH-THFで脱シリル化して (2-2) を得た。
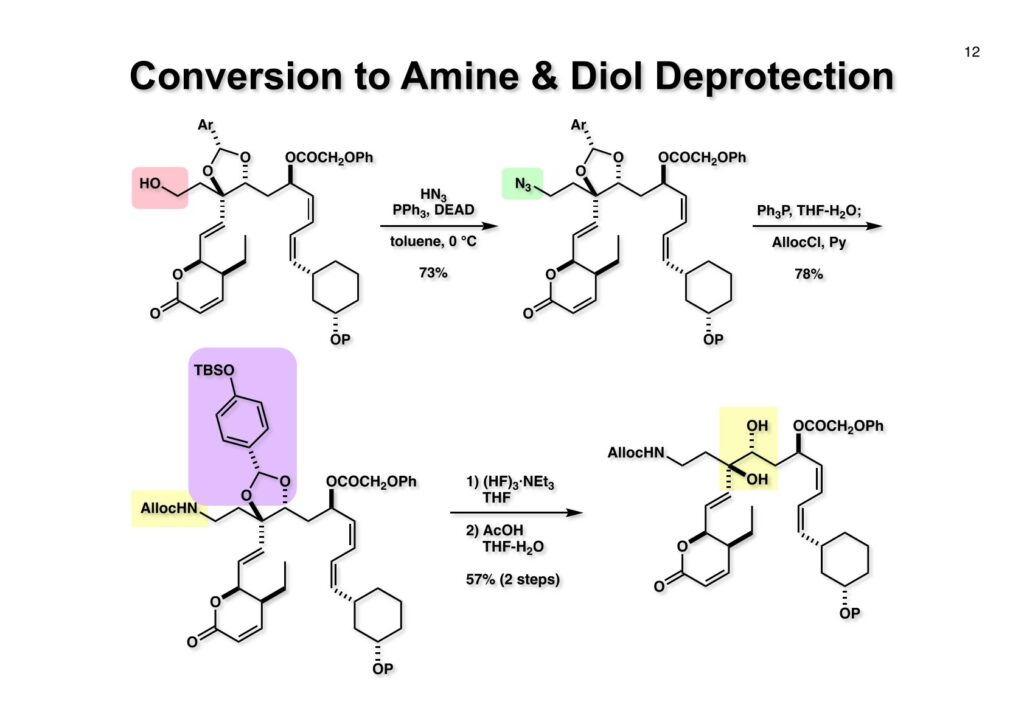 アルコールを光延反応でアジドに変換するのにPPTSを使うのが一般的だが、私は断然HN3を発生させ、そのトルエン溶液を使う。とにかく圧倒的に反応が速いのだ。光延反応に使うHN3のトルエン溶液の製法は、まずNaN3のトルエン懸濁液に水を少量加え、次に濃硫酸を滴下する。そして脱水のためNa2SO4を加えれば出来上がりである。(1-1) とHN3の光延反応は0 °C10分間で終了して (1-2) が得られた。アジドからアミンへの変換はPh3Pを使うStaudinger反応で行い、反応溶液にAllocCl-Pyを加えてAlloc化し (2-1) を得た。ここでリン酸基を導入するためにアセタールを除去することにした。まず弱いフッ素源である(HF)3·NEt3でTBS基を外し、酢酸-THF-水を用いて室温放置するとジオール (2-2) が得られた。
アルコールを光延反応でアジドに変換するのにPPTSを使うのが一般的だが、私は断然HN3を発生させ、そのトルエン溶液を使う。とにかく圧倒的に反応が速いのだ。光延反応に使うHN3のトルエン溶液の製法は、まずNaN3のトルエン懸濁液に水を少量加え、次に濃硫酸を滴下する。そして脱水のためNa2SO4を加えれば出来上がりである。(1-1) とHN3の光延反応は0 °C10分間で終了して (1-2) が得られた。アジドからアミンへの変換はPh3Pを使うStaudinger反応で行い、反応溶液にAllocCl-Pyを加えてAlloc化し (2-1) を得た。ここでリン酸基を導入するためにアセタールを除去することにした。まず弱いフッ素源である(HF)3·NEt3でTBS基を外し、酢酸-THF-水を用いて室温放置するとジオール (2-2) が得られた。
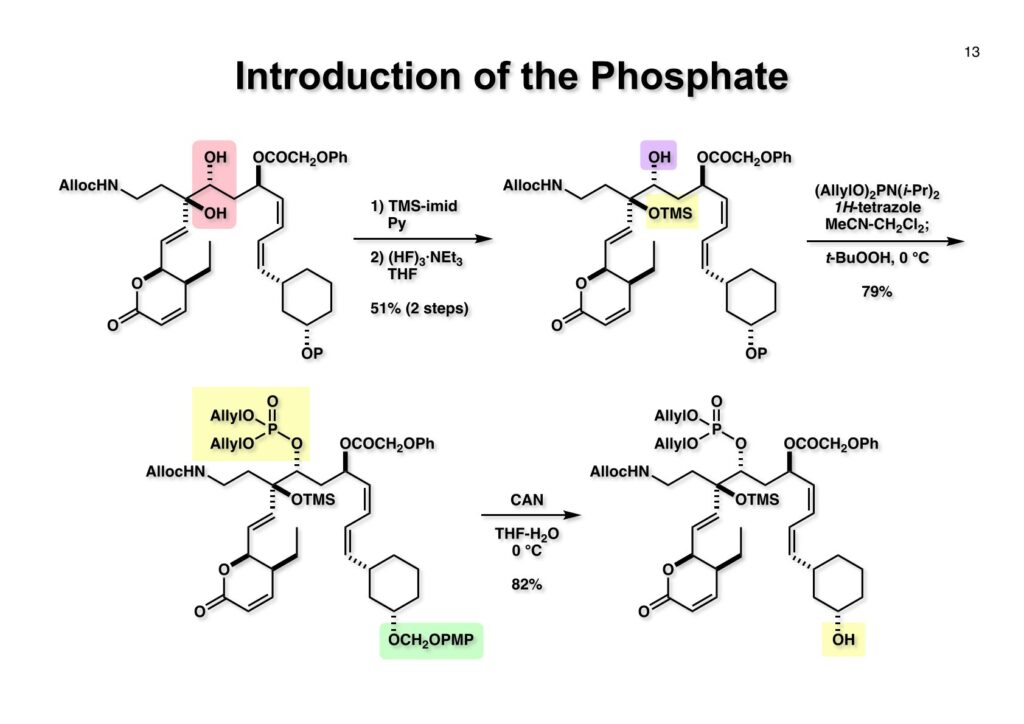 ジオール (1-1) の2級水酸基を選択的にリン酸化しようとしたが3級水酸基もリン酸化されたりする混合物が生成したため、TMS化されたイミダゾールを使ってジオールをTMS化してから、(HF)3·NEt3を用いて2級アルコールのTMS基だけを外して (1-2) を得た。これを核酸合成で常用される亜リン酸エステル化してt-BuOOHで酸化する方法でリン酸エステル (2-1) に変換した。次いでCAN酸化によって (2-1) のPMPOCH2基を除去してアルコール (2-2) を得た。
ジオール (1-1) の2級水酸基を選択的にリン酸化しようとしたが3級水酸基もリン酸化されたりする混合物が生成したため、TMS化されたイミダゾールを使ってジオールをTMS化してから、(HF)3·NEt3を用いて2級アルコールのTMS基だけを外して (1-2) を得た。これを核酸合成で常用される亜リン酸エステル化してt-BuOOHで酸化する方法でリン酸エステル (2-1) に変換した。次いでCAN酸化によって (2-1) のPMPOCH2基を除去してアルコール (2-2) を得た。
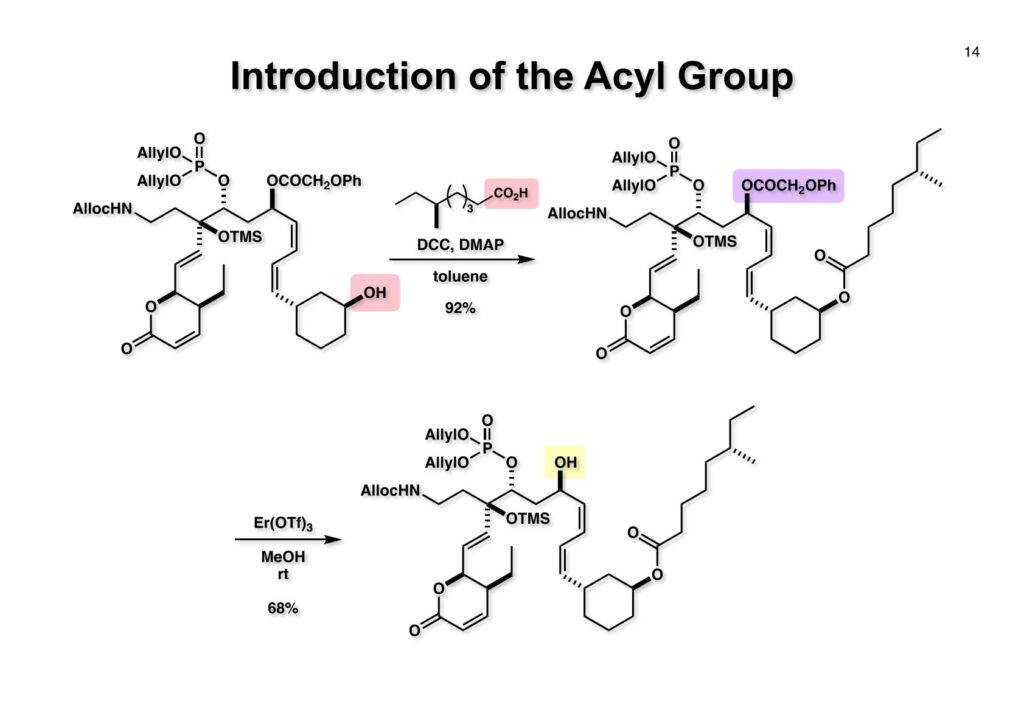 (1-1) の水酸基と(S)-6-methyloctanoic acid (1-2) をDCC-DMAPを用いて縮合し (1-3) を得た。(1-3) のフェノキシアセテートの除去条件の検討を行ったが、酸性条件はもちろん使えないが、塩基性条件も副反応が生じた。以前、光学活性ゲルセミンの全合成の際にEvansの不斉補助基の除去にメタノール中Sm(OTf)3を用いて高収率でメチルエステルを得たことがあるのでランタナイドトリフレートを試したところEr(OTf)3がまずまずの結果を与えた。(2-1) を68%の収率で得て14%の出発物 (1-3) を回収した。フェノキシ基の酸素にエルビウムが配位して活性化してsolvolysisが進行していると思われるので、試してはいないがメトキシアセテートにしておいたら好結果が得られたかもしれない。
(1-1) の水酸基と(S)-6-methyloctanoic acid (1-2) をDCC-DMAPを用いて縮合し (1-3) を得た。(1-3) のフェノキシアセテートの除去条件の検討を行ったが、酸性条件はもちろん使えないが、塩基性条件も副反応が生じた。以前、光学活性ゲルセミンの全合成の際にEvansの不斉補助基の除去にメタノール中Sm(OTf)3を用いて高収率でメチルエステルを得たことがあるのでランタナイドトリフレートを試したところEr(OTf)3がまずまずの結果を与えた。(2-1) を68%の収率で得て14%の出発物 (1-3) を回収した。フェノキシ基の酸素にエルビウムが配位して活性化してsolvolysisが進行していると思われるので、試してはいないがメトキシアセテートにしておいたら好結果が得られたかもしれない。
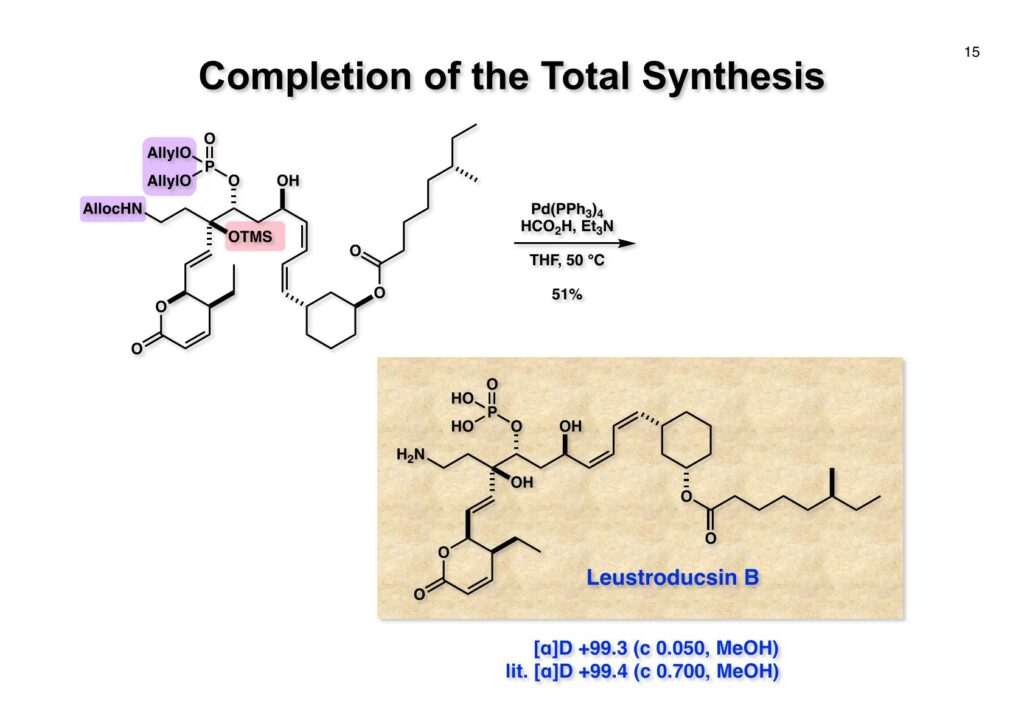 最終段階は (1-1) 中の3個のアリル基をPd触媒とギ酸-Et3Nを用いて除去する反応で、THF中50 °Cで加熱するうちにTMS基も除去されてleustroducsin B (2-1) が得られて全合成は完了した。旋光度も標準標品と良い一致を示した。私としてはちょっと毛色の違う化合物を、島田さんや鏑木君と試行錯誤を重ねながら経験を積んでいった教育的な研究だったと言える。
最終段階は (1-1) 中の3個のアリル基をPd触媒とギ酸-Et3Nを用いて除去する反応で、THF中50 °Cで加熱するうちにTMS基も除去されてleustroducsin B (2-1) が得られて全合成は完了した。旋光度も標準標品と良い一致を示した。私としてはちょっと毛色の違う化合物を、島田さんや鏑木君と試行錯誤を重ねながら経験を積んでいった教育的な研究だったと言える。