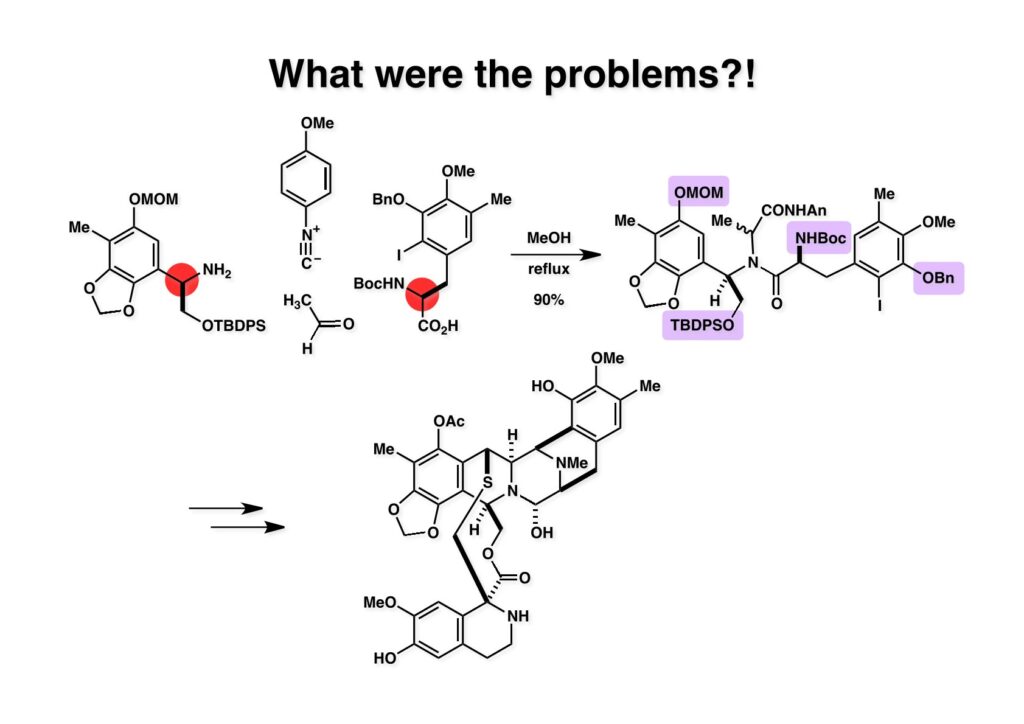
Ecteinascidin 743 (ET-743) はカリブ海の群体ホヤから単離された抗腫瘍活性を持つ海洋性天然物でイリノイ大学のKen Rinehart教授が構造決定した。北大の酒井隆一教授はRinehart研の大学院生として単離構造決定に参加された。スペインのPharmaMarという会社からYondelisという商品名で主に軟部肉腫の治療薬として使用されている。私がライス大学に居た時にPharmaMarから全合成研究をしてほしいと依頼され、safaramycin系化合物の親玉のように複雑な構造を有するところに興味を持って全合成に取りかかった。程なくして東大に移ることになり3万ドル余りあった研究費をライス大学から東大に持ってこようとしたら、外国の会社の金を研究費として移管するなど出来ないと東大の事務に言われたのでPharmaMarに返還した経緯がある。東大では助手の菅さんの下につけた学部4年の遠藤篤史君(横島君の同級生)に、特許を出さないといけない研究だから学会発表は出来ないけど、博士号を取るまでには必ず全合成を終わらせるようにと含みを持たせて任せることにした。全合成論文に名前が連なる連中は最終段階で助っ人に加わった柳沢さん以外は加勢を多少した程度で、95%は遠藤君の努力が成し遂げた研究である。その後第二世代の全合成は、横島准教授の下で3名の院生(乾朋彦君、河岸文希君、藤間達哉君)の合作として完成させた。全合成研究で難しいのは誰がどのような貢献をしたか、ということで、運悪く袋小路に追い込まれて日の目を見なかったアプローチも多々あり、それはやっていた研究者の運不運とか技量にも関わるし、また上の者(つまり私)のアイデアがトロかったということもあり、で研究に関わった全員の名前を平等に列挙出来ないのが心苦しいところである(論文には載せるけれど)。
“Total Synthesis of Ecteinascidin 743,” A. Endo, A. Yanagisawa, M. Abe, S. Tohma, T. Kan, and T. Fukuyama, J. Am. Chem. Soc. , 124 , 6552 (2002).
“Total Synthesis of Ecteinascidin 743,” F. Kawagishi, T. Toma, T. Inui, S. Yokoshima, and T. Fukuyama, J. Am. Chem. Soc. , 135 , 13684-13687 (2013).
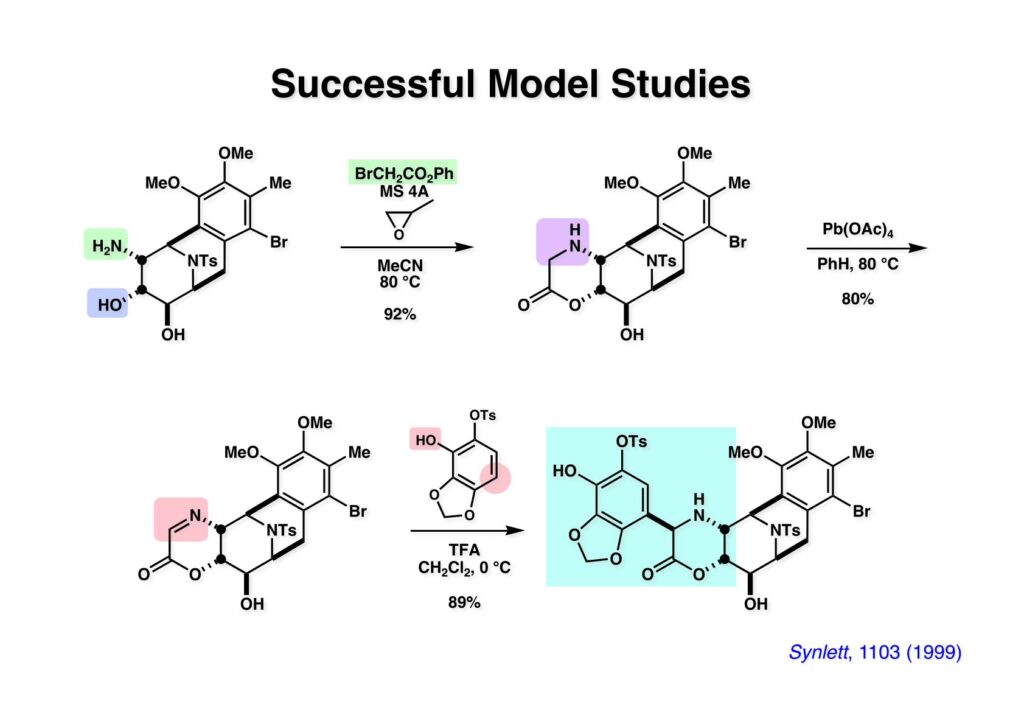
“Synthetic Study on Ecteinascidin 743 Starting from D-Glucose,” A. Endoh, T. Kan, and T. Fukuyama, Synlett , 1103 (1999); “Synthetic Studies toward Ecteinascidin 743 (Trabectedin),” T. Imai, H. Nakata, S. Yokoshima, and T. Fukuyama, Synthesis , 44 , 2743-2753 (2012).
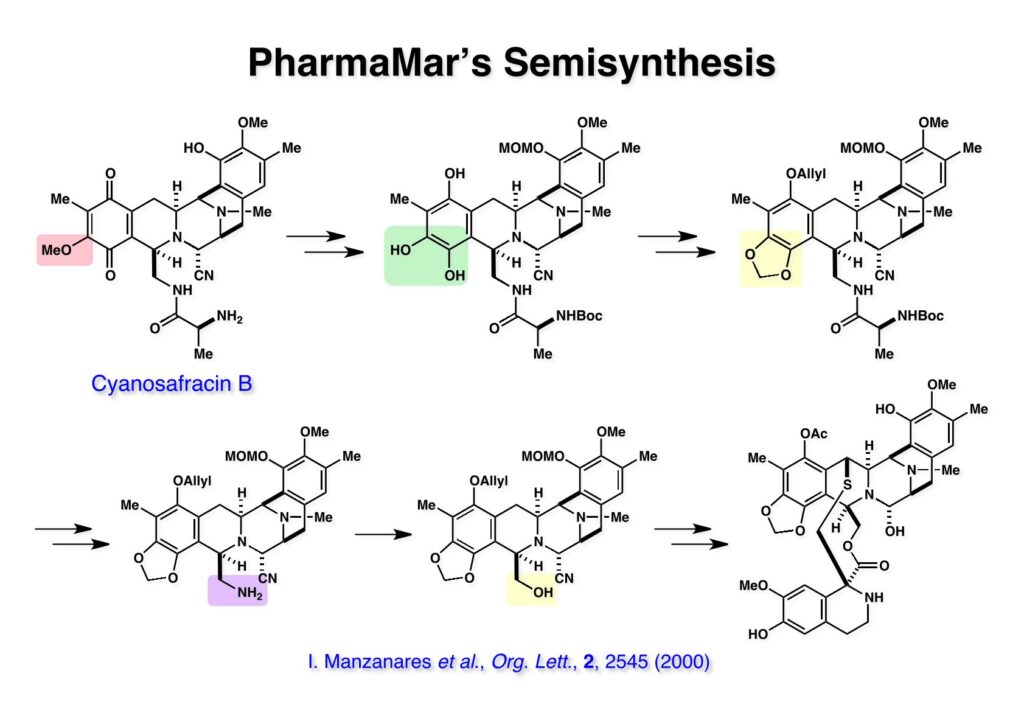
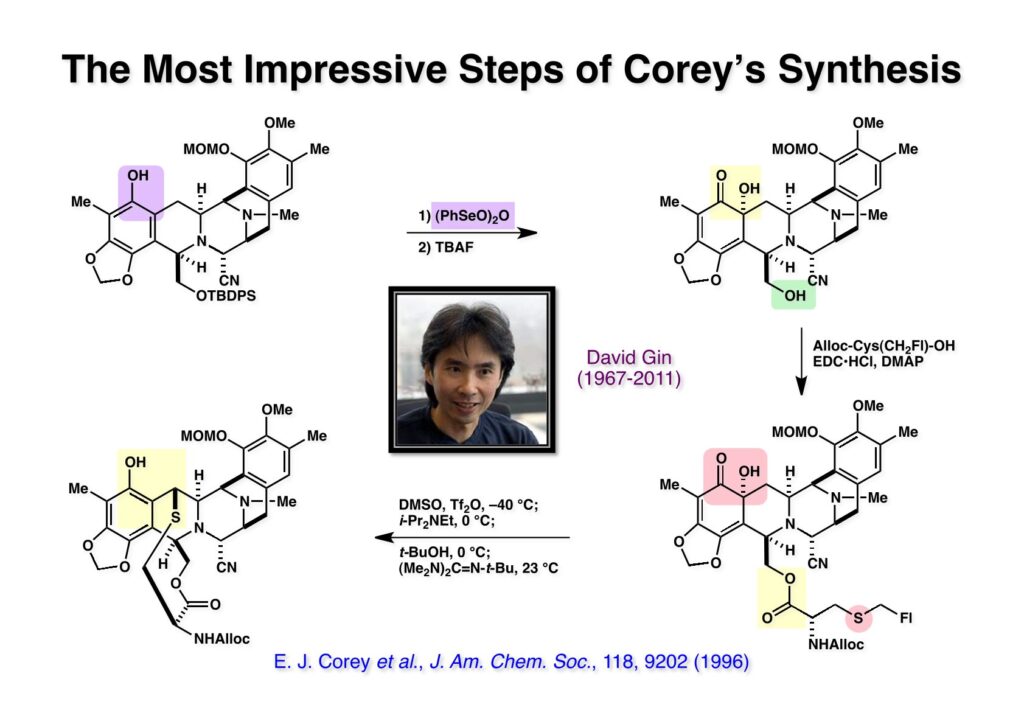
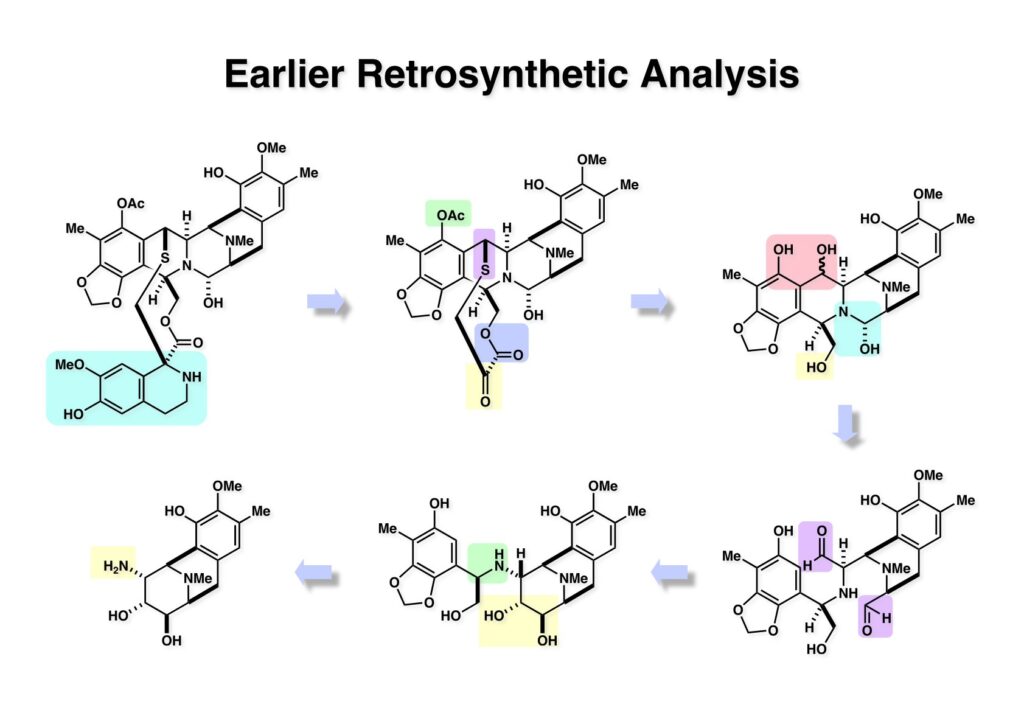
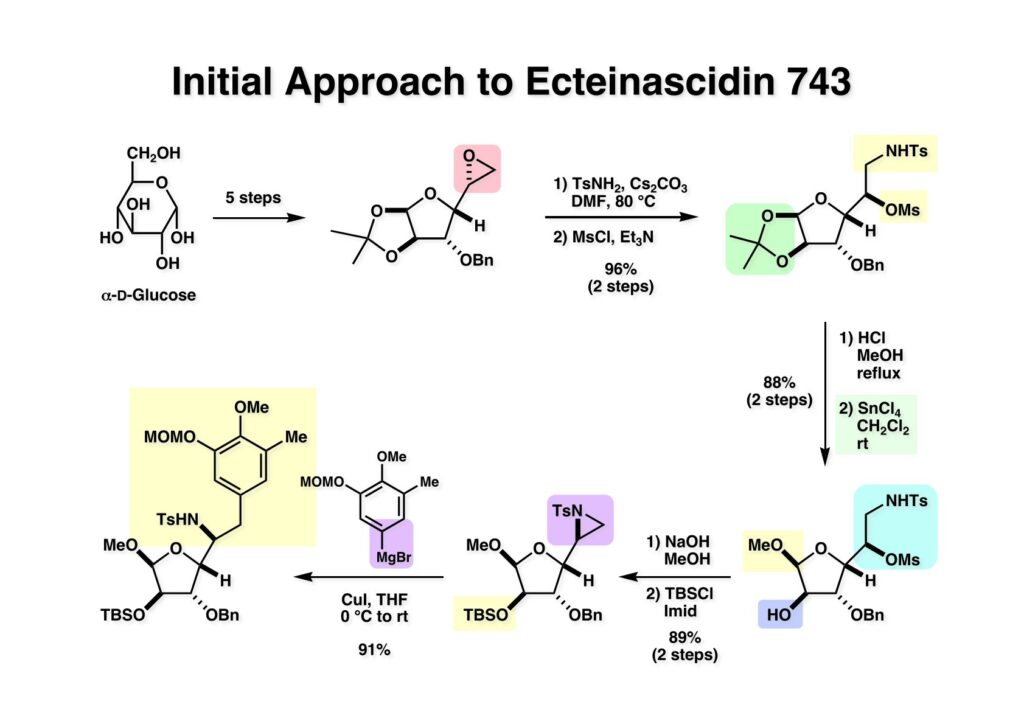
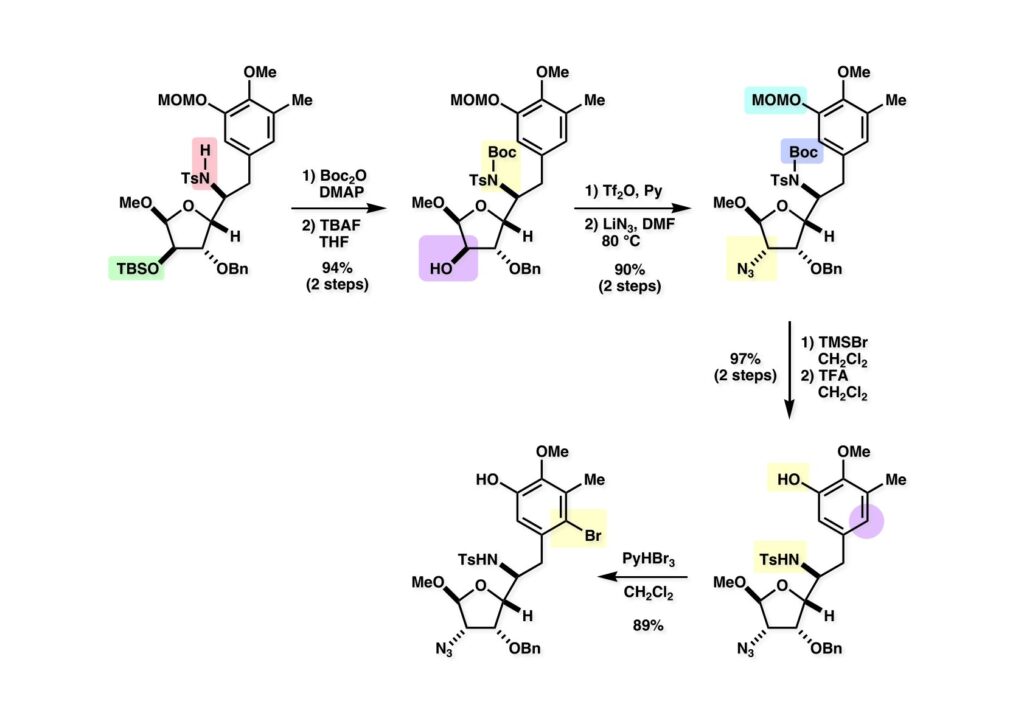
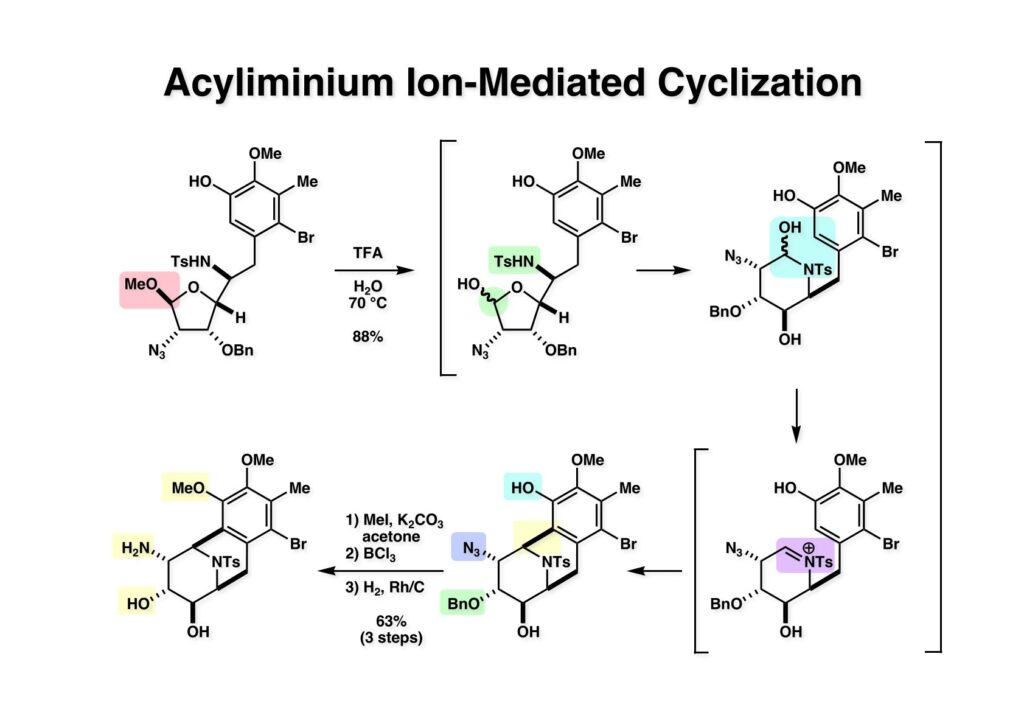
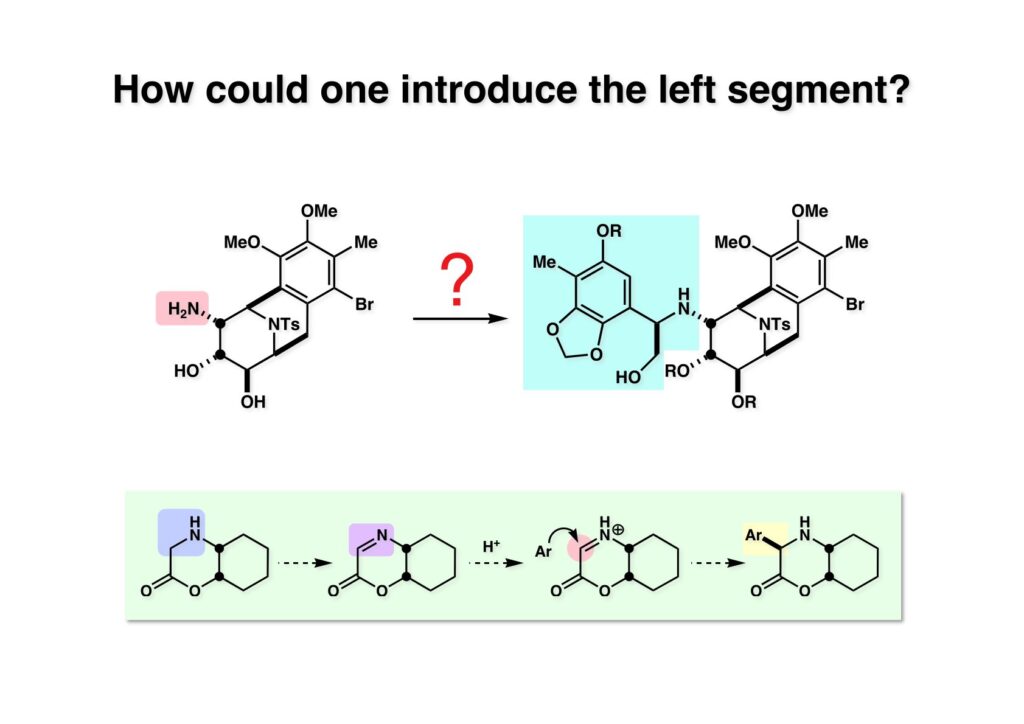
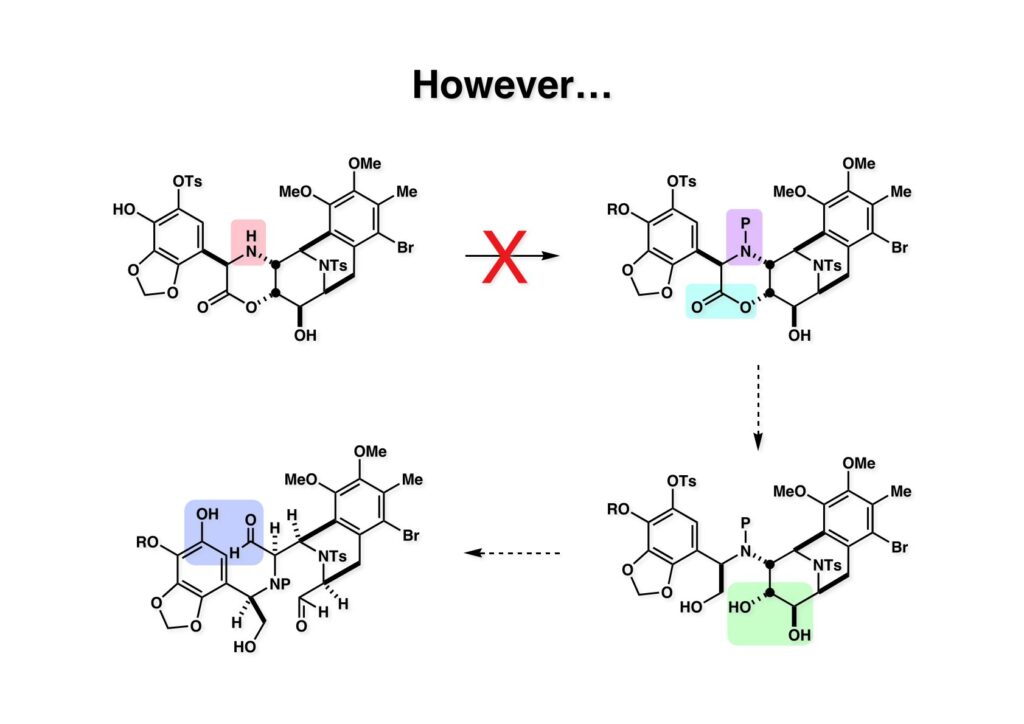
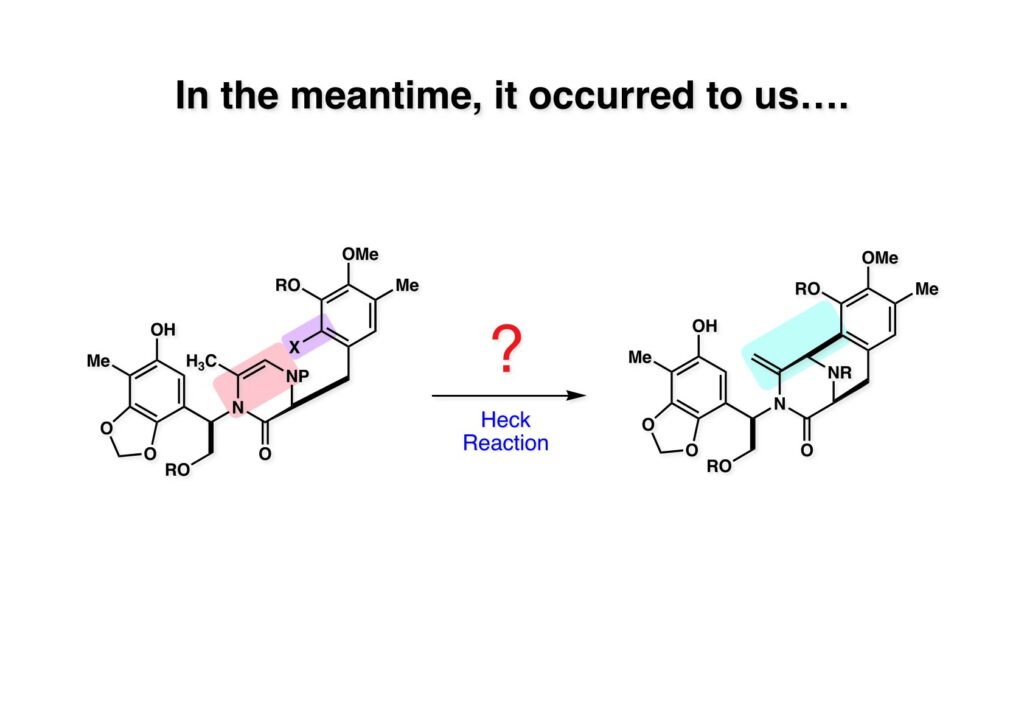
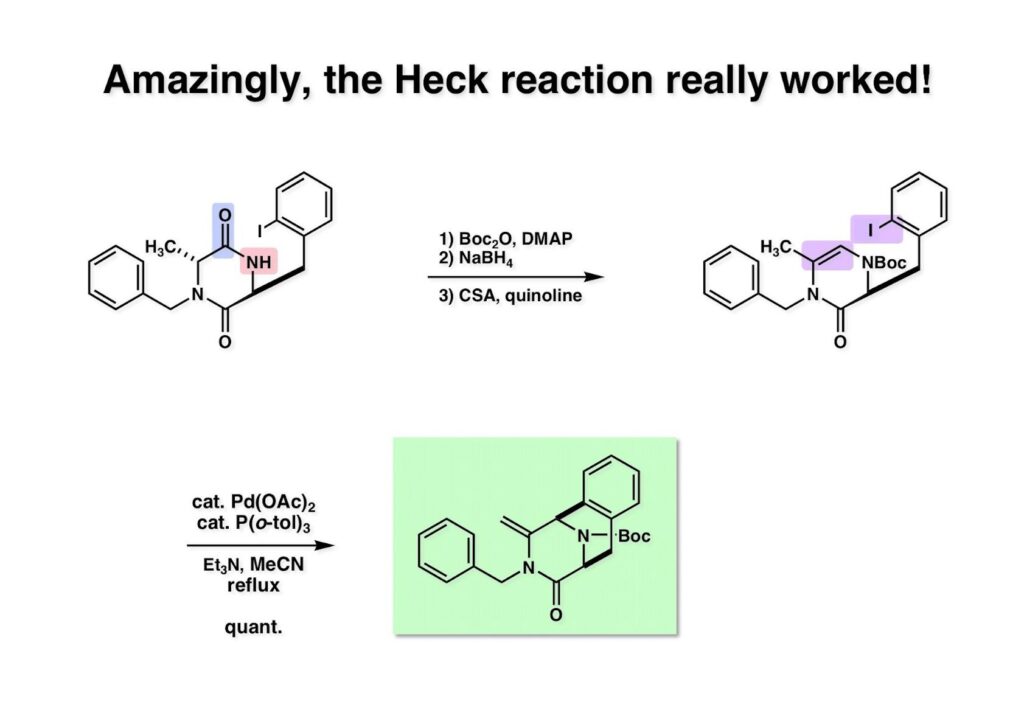
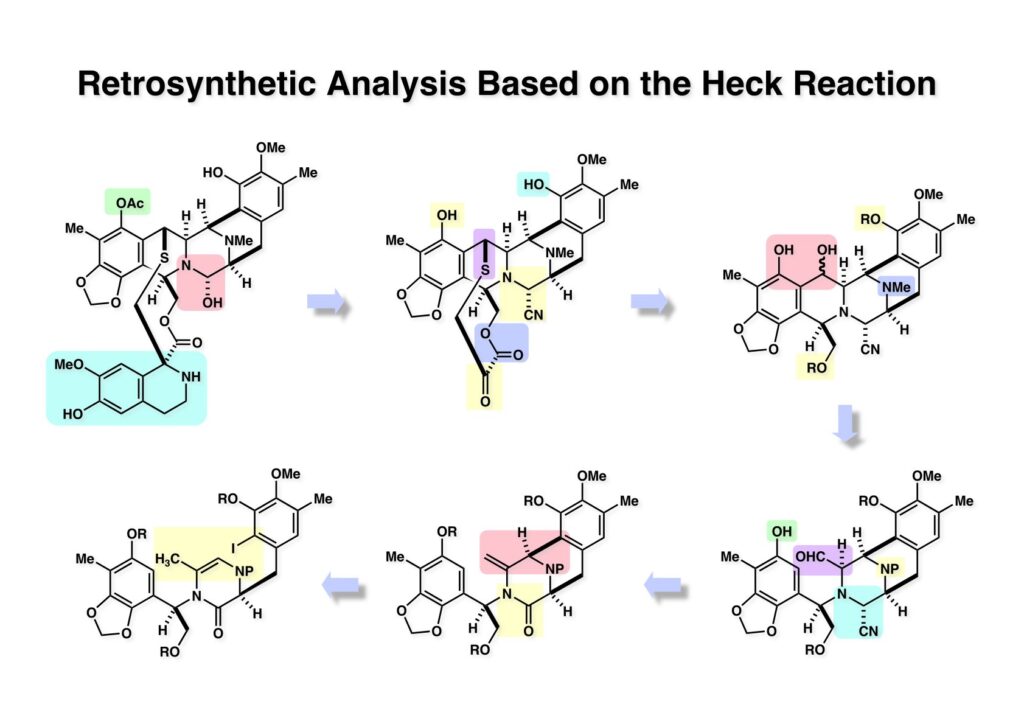
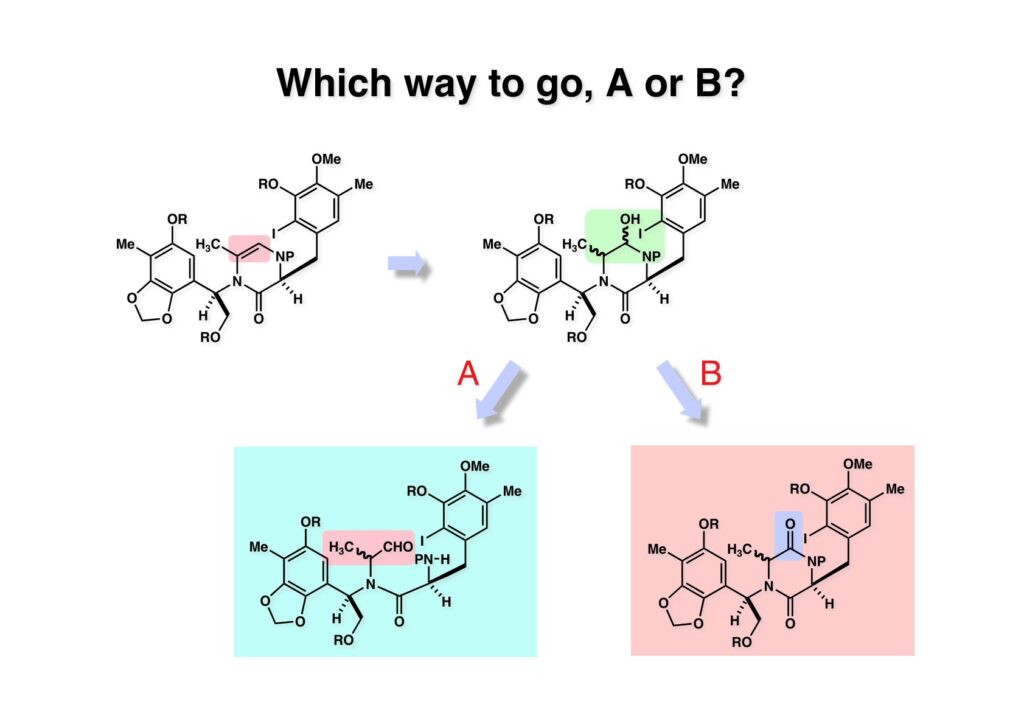
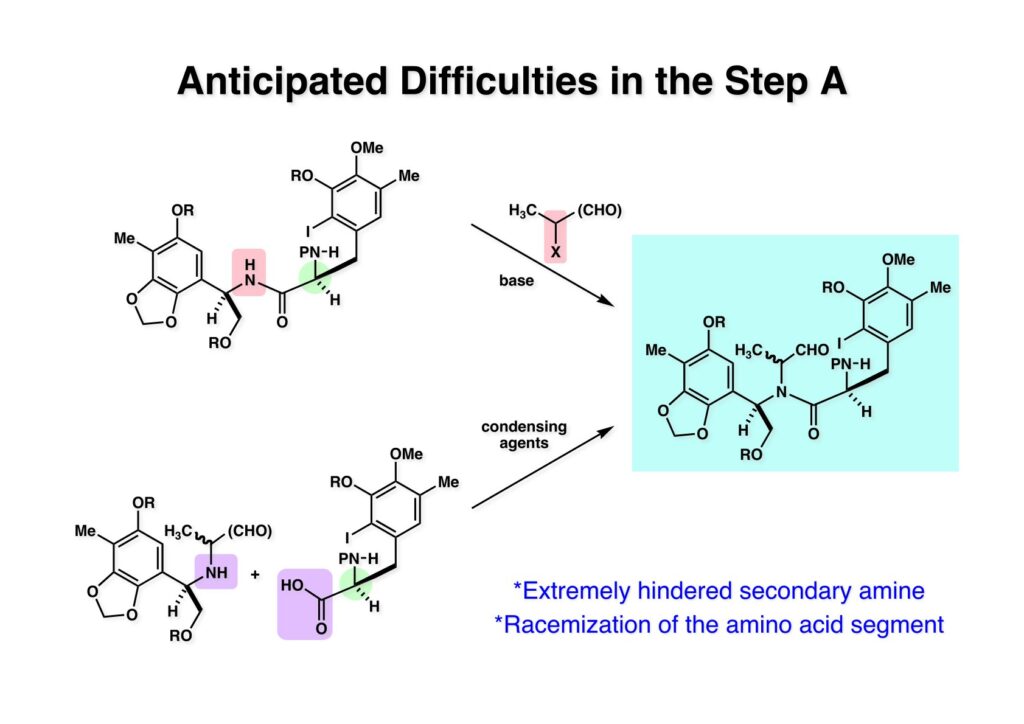
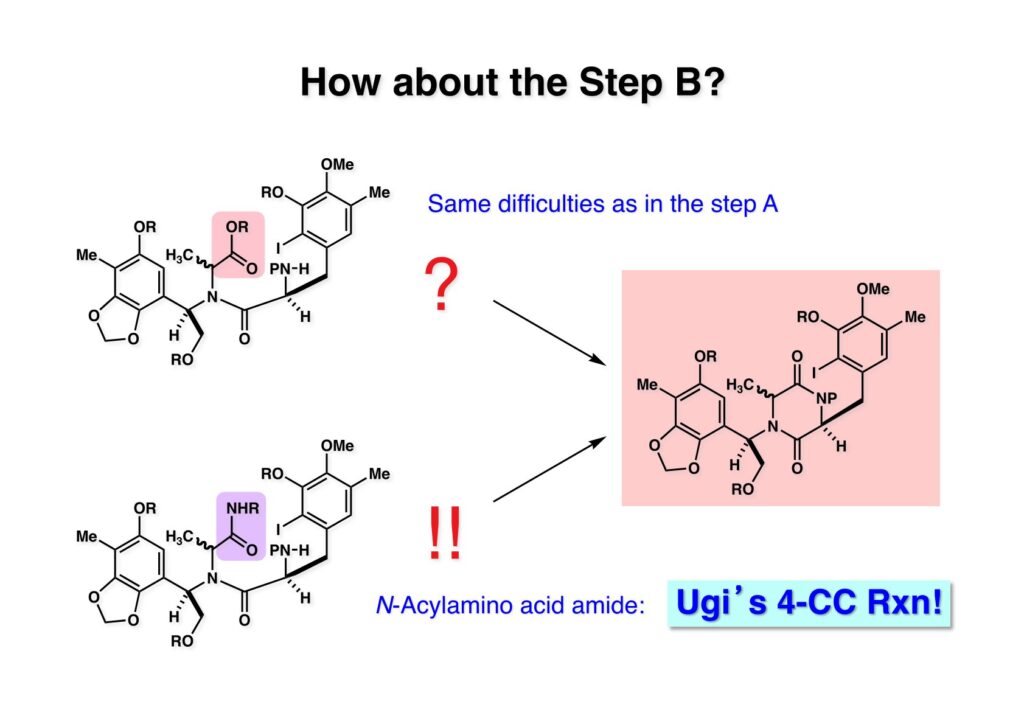
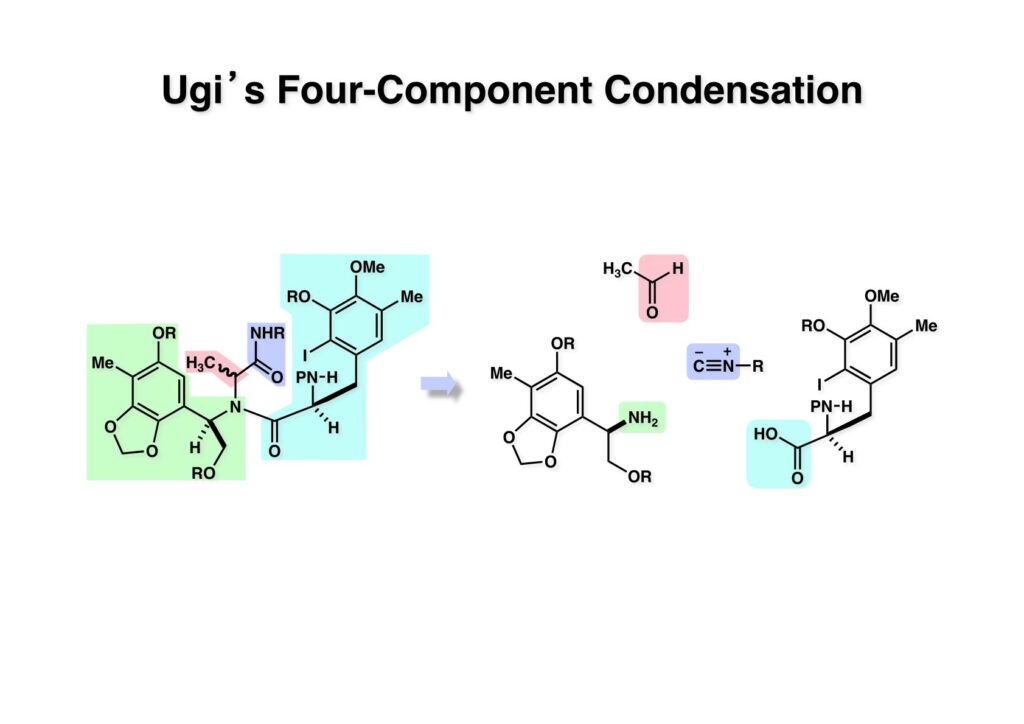
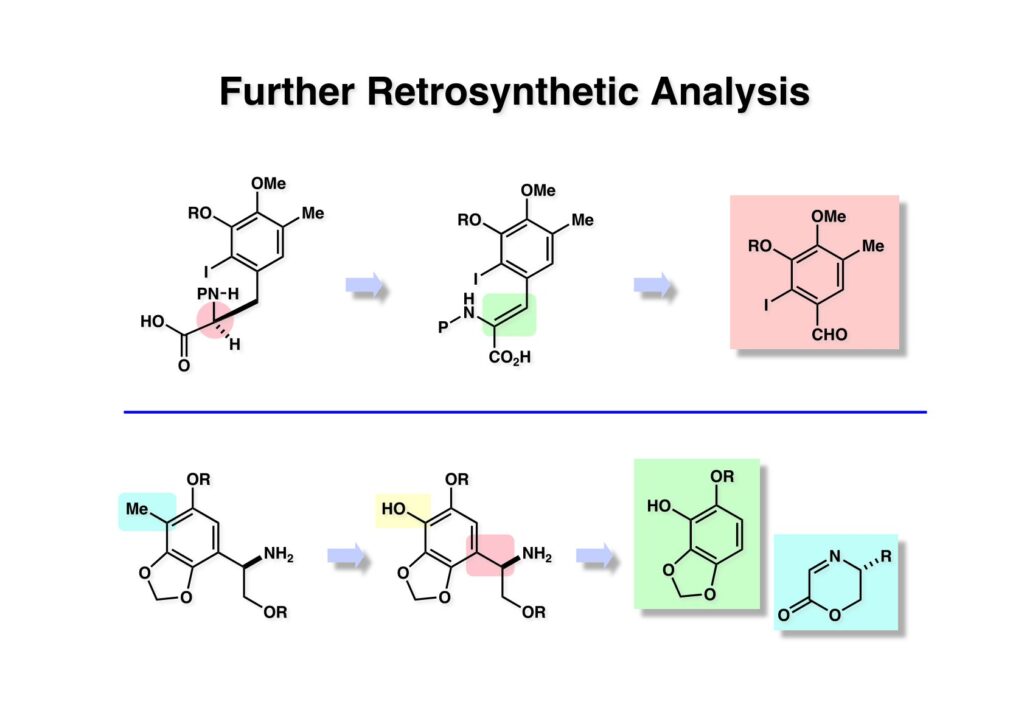
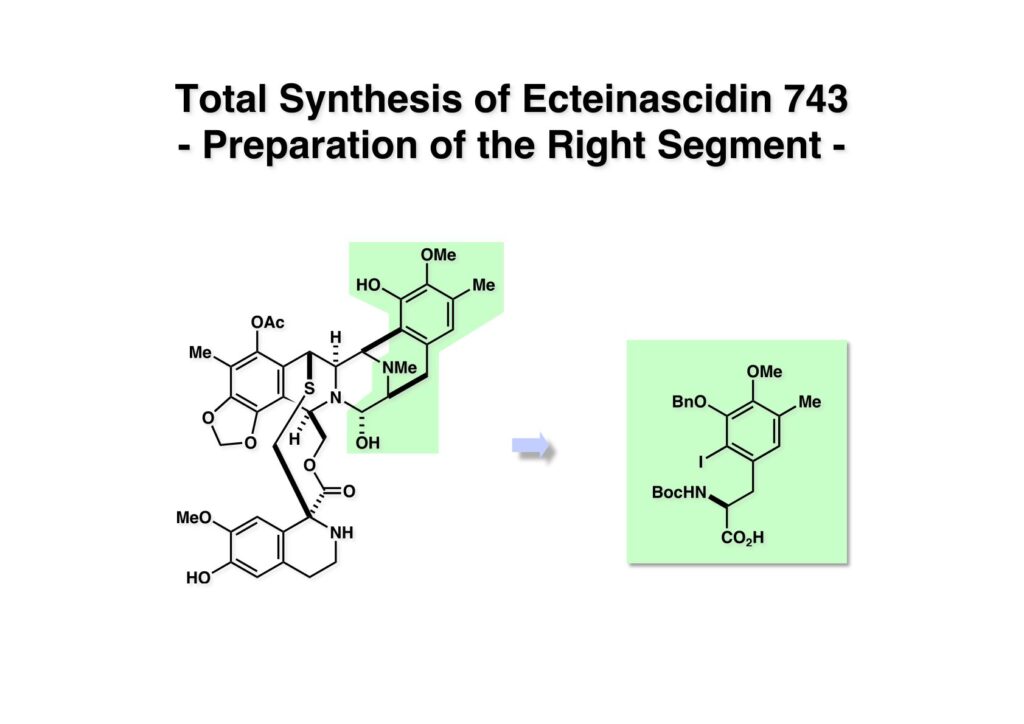
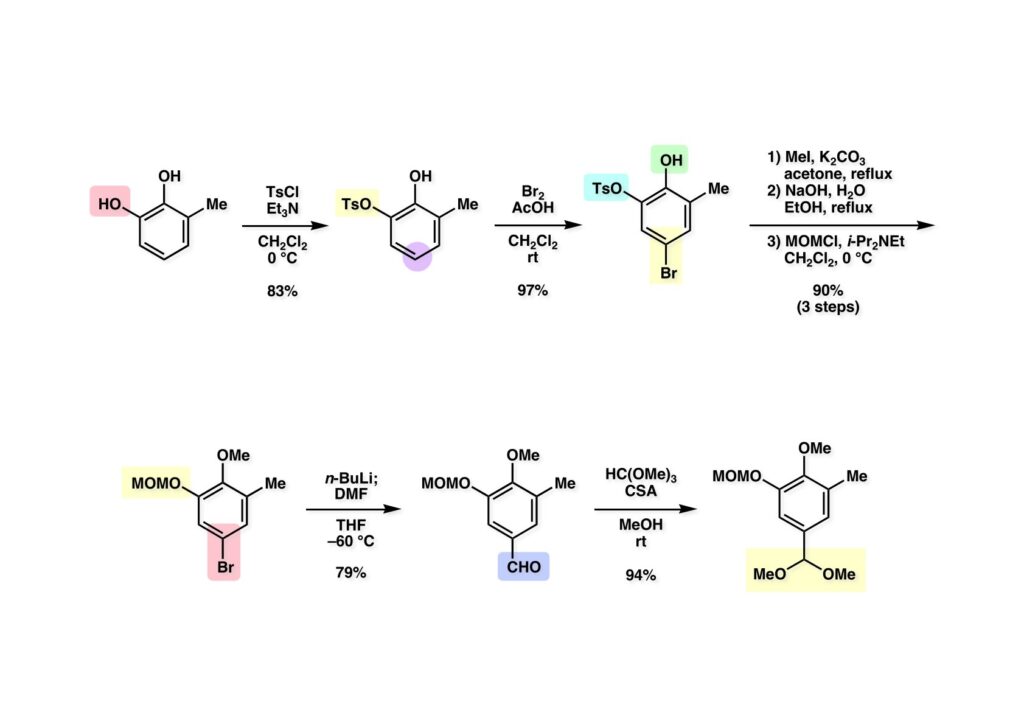
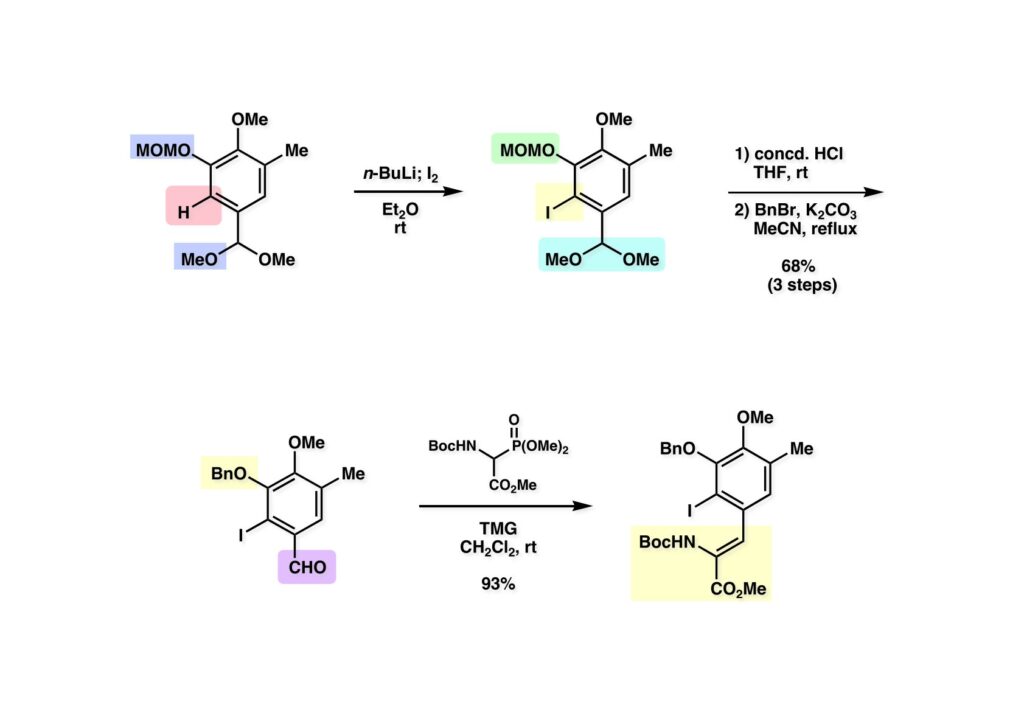
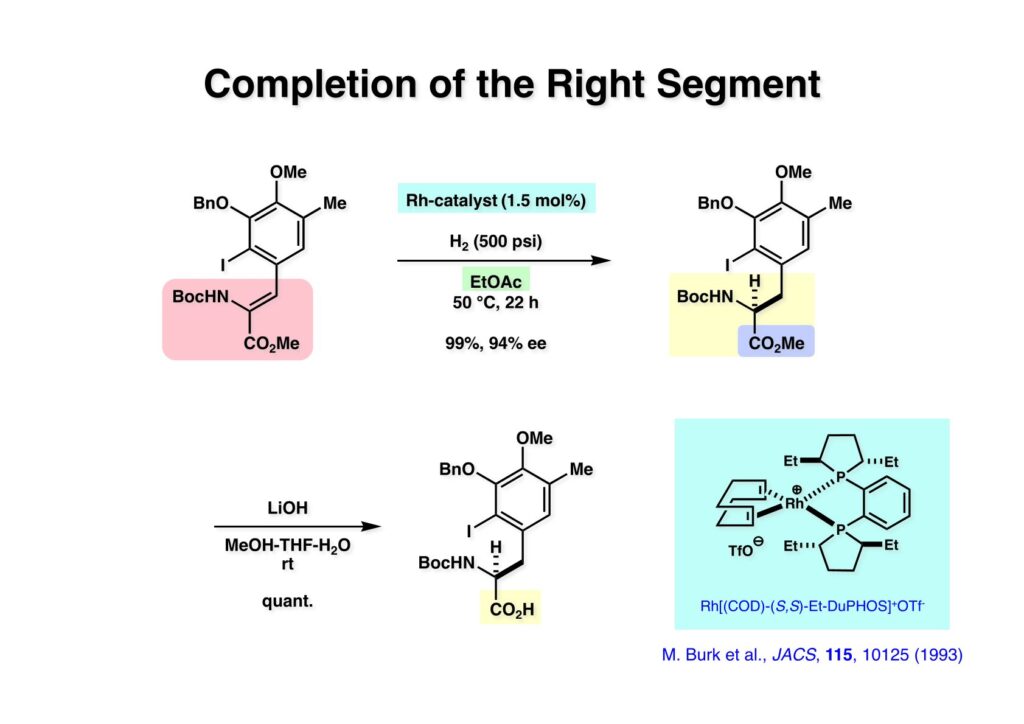
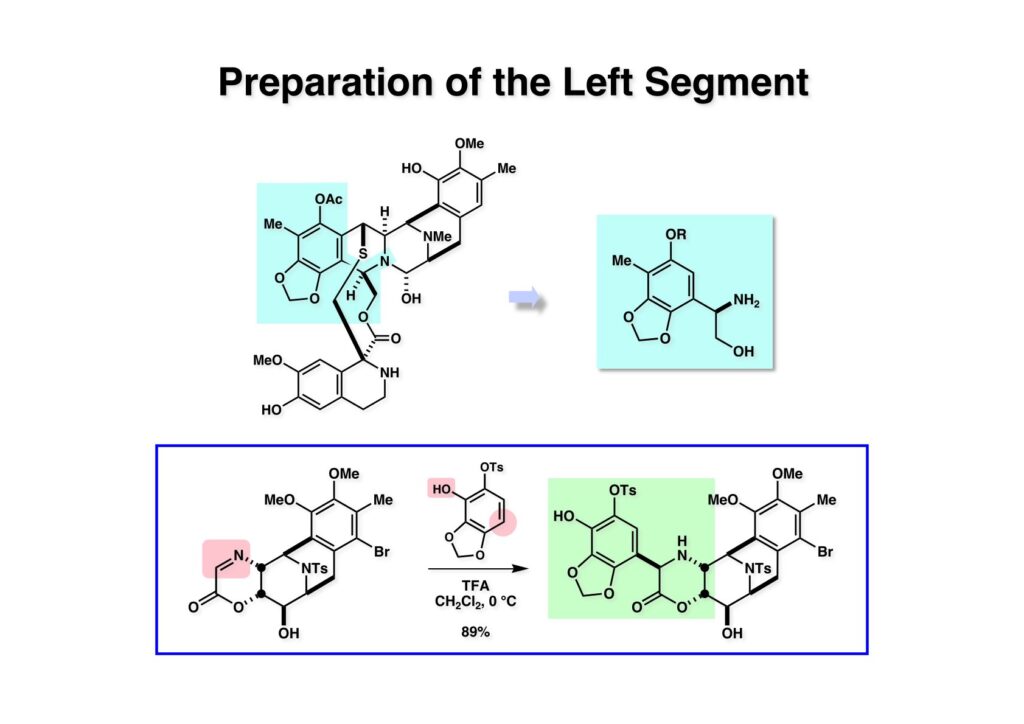
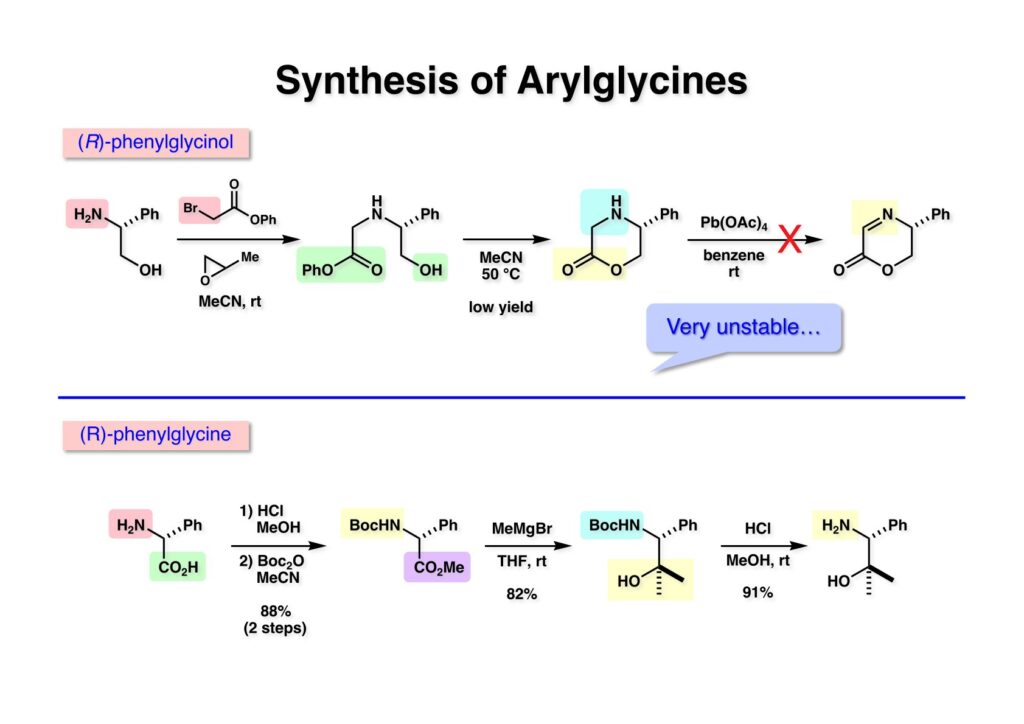
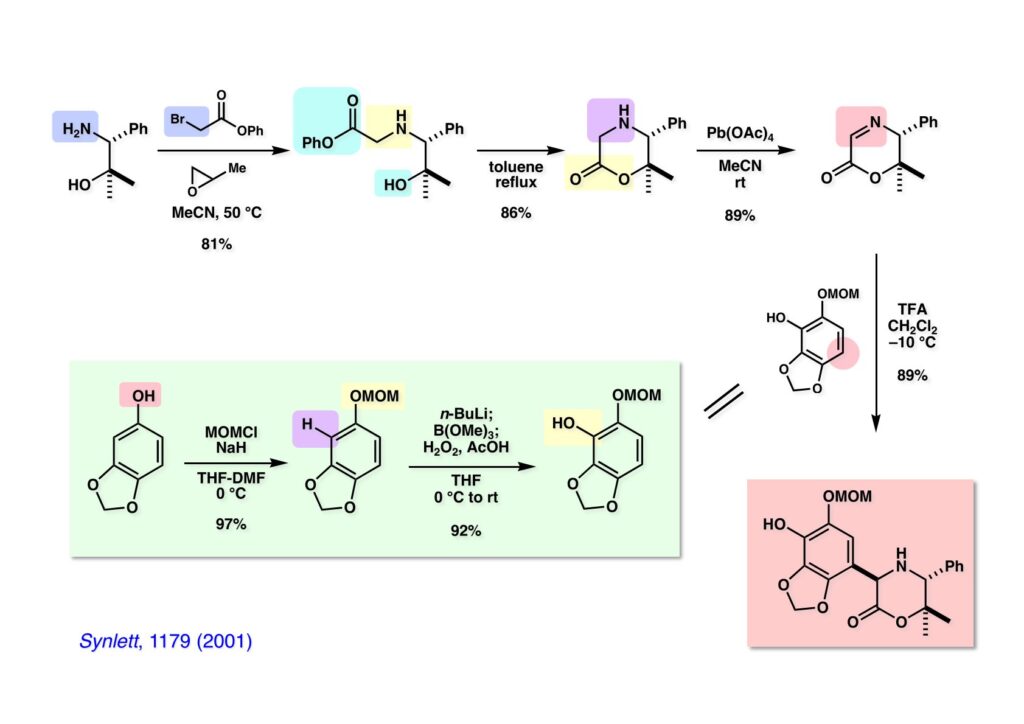
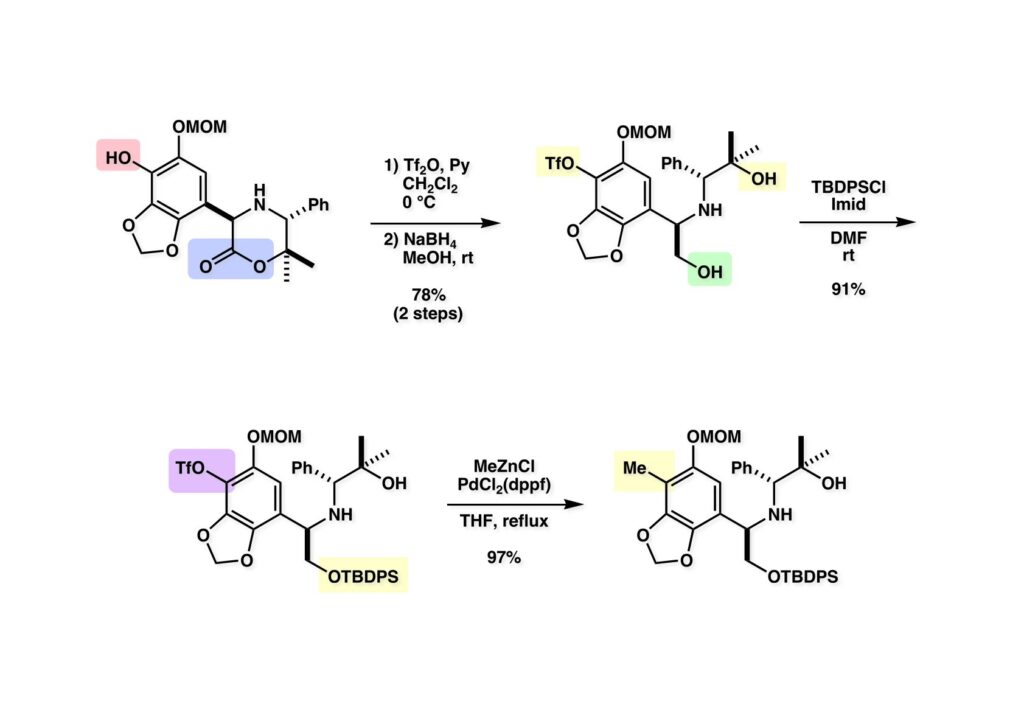
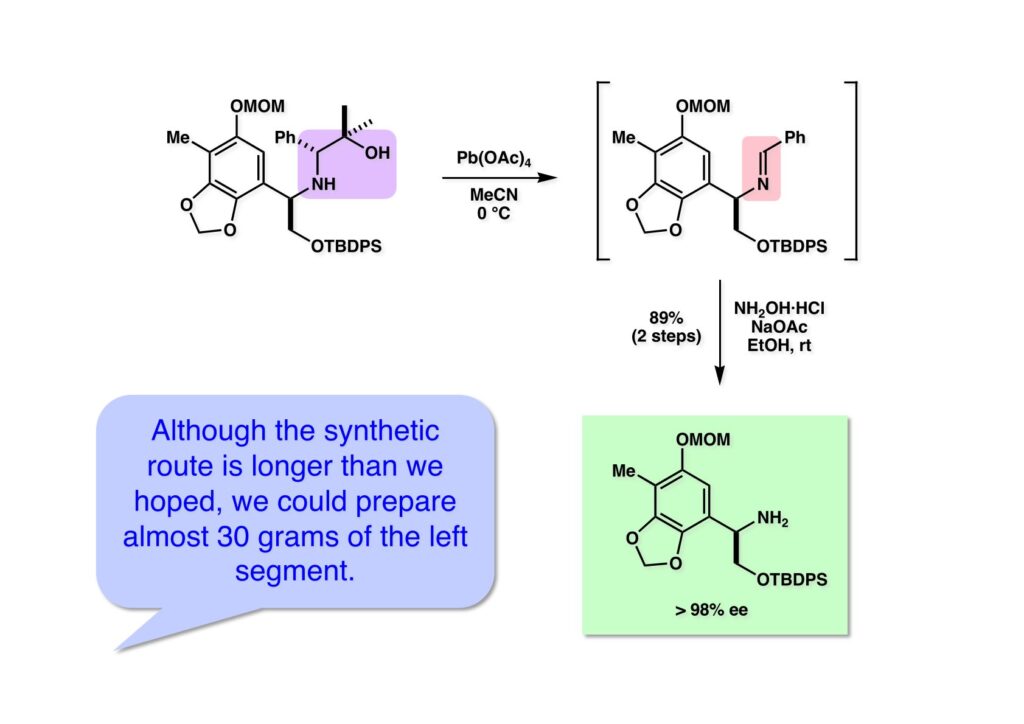
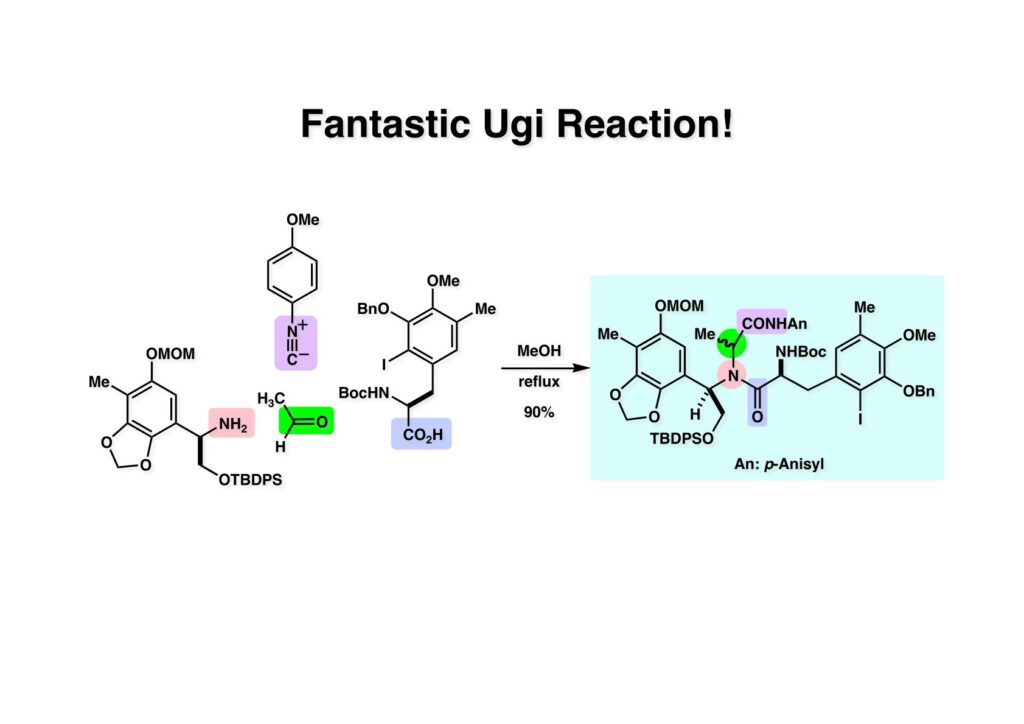
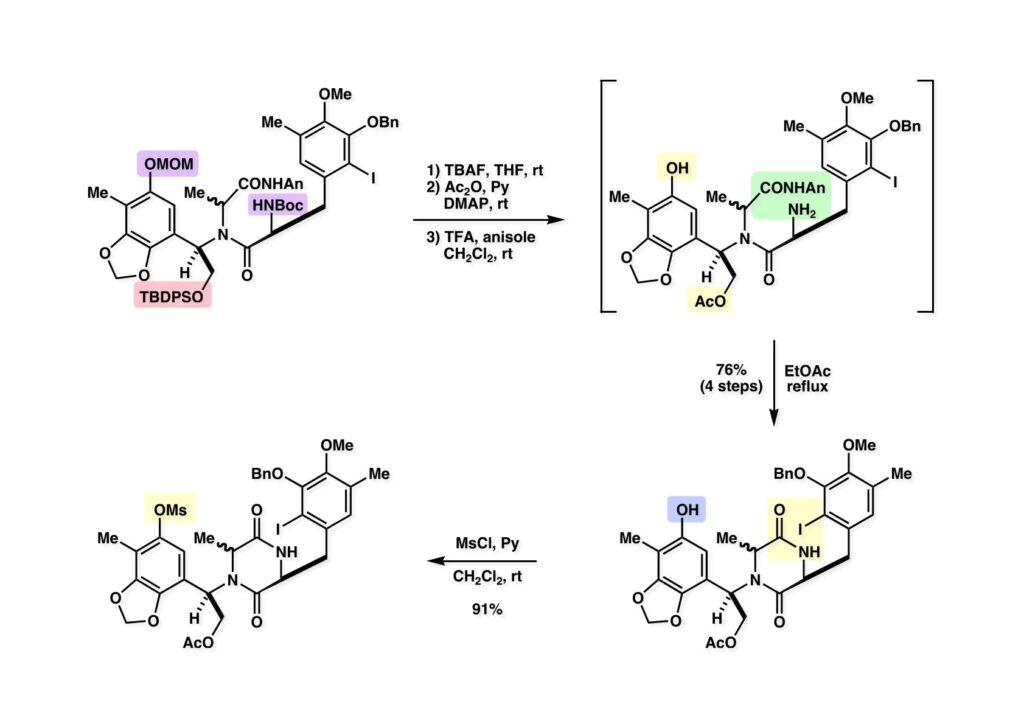
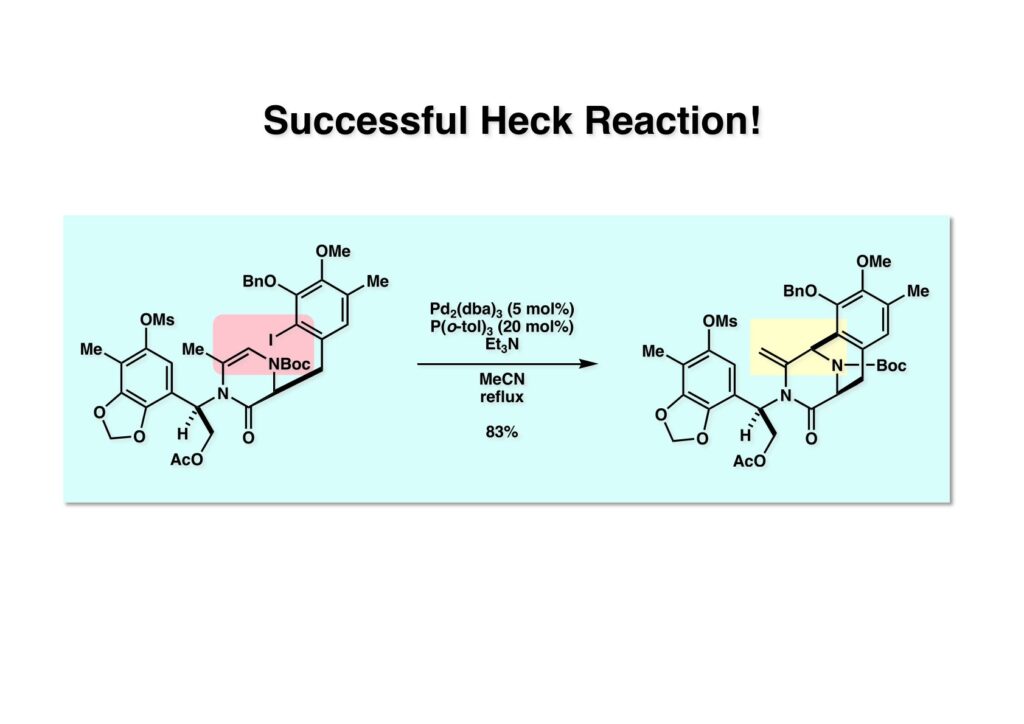
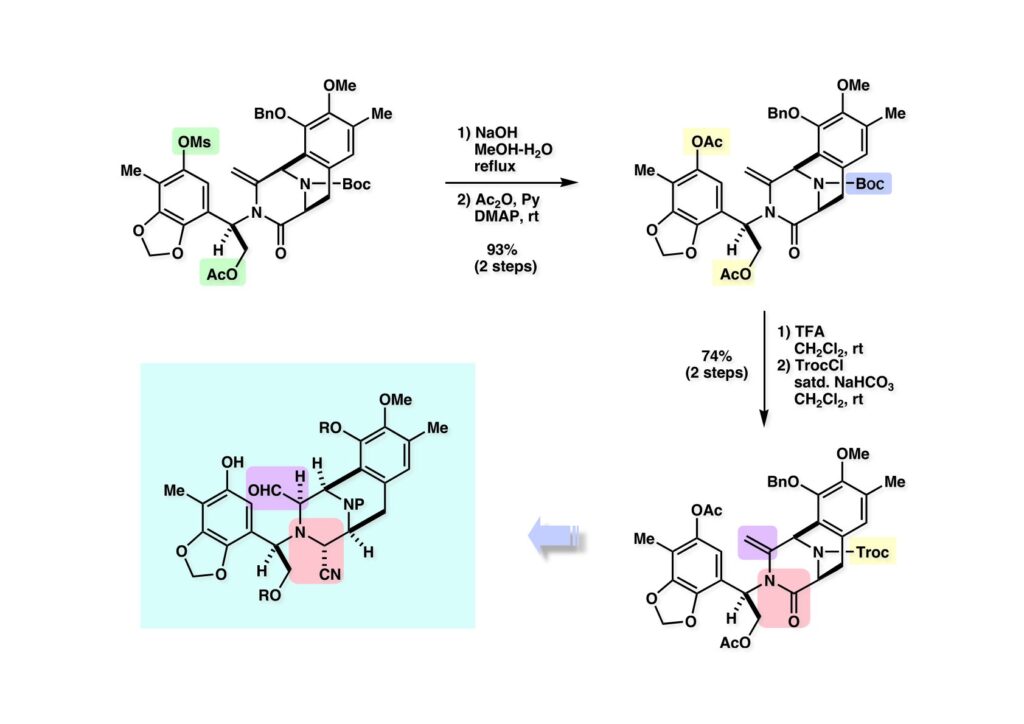
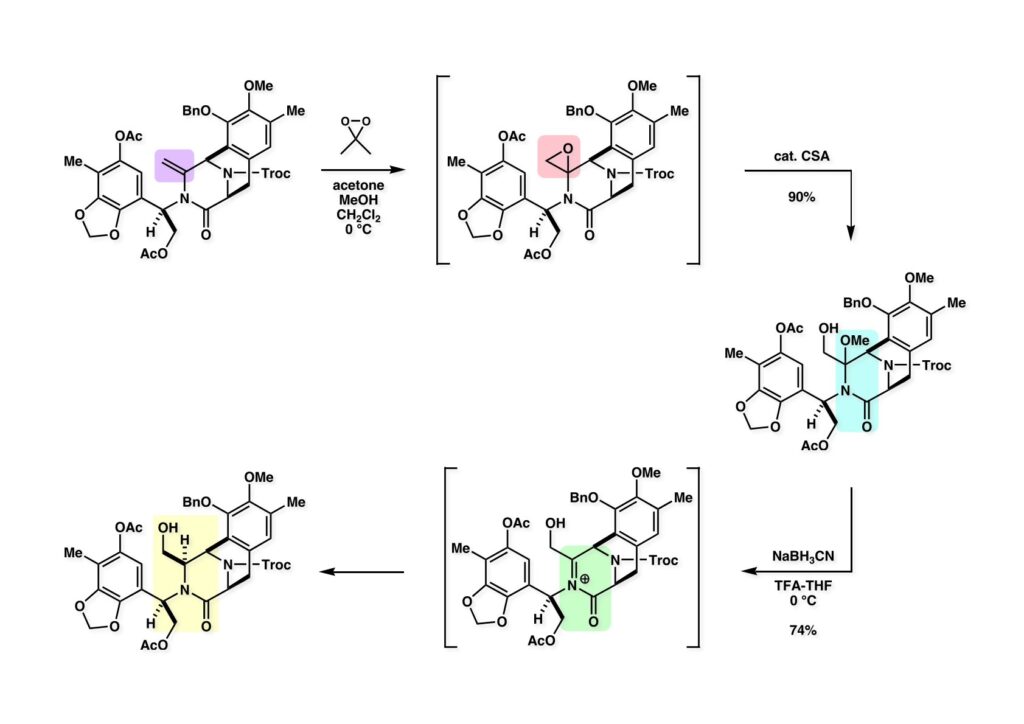
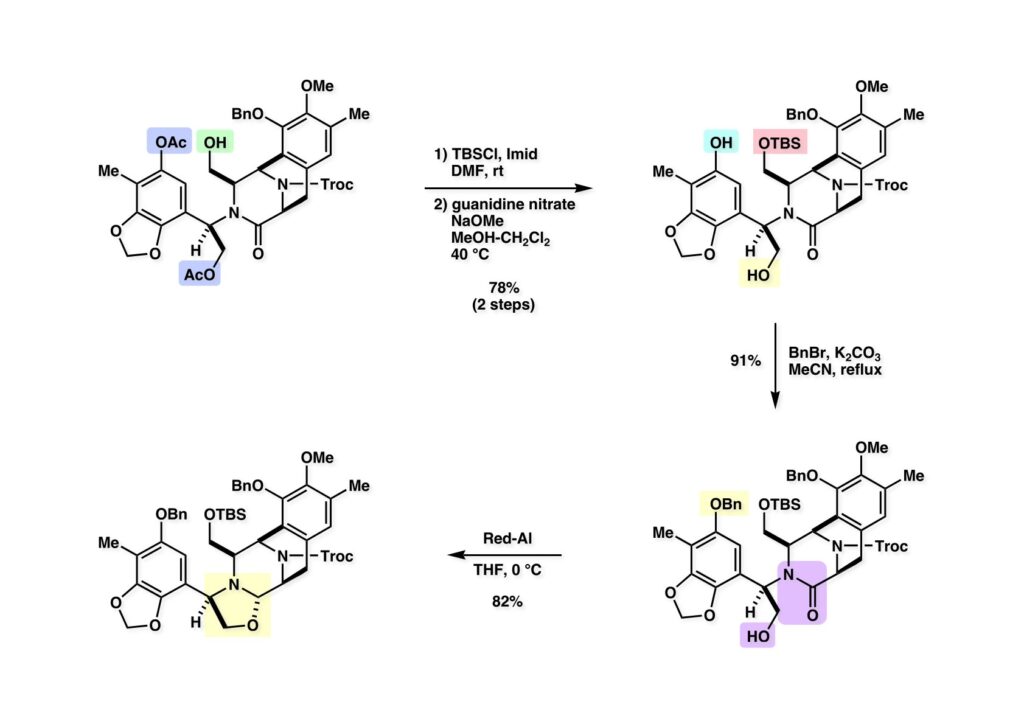
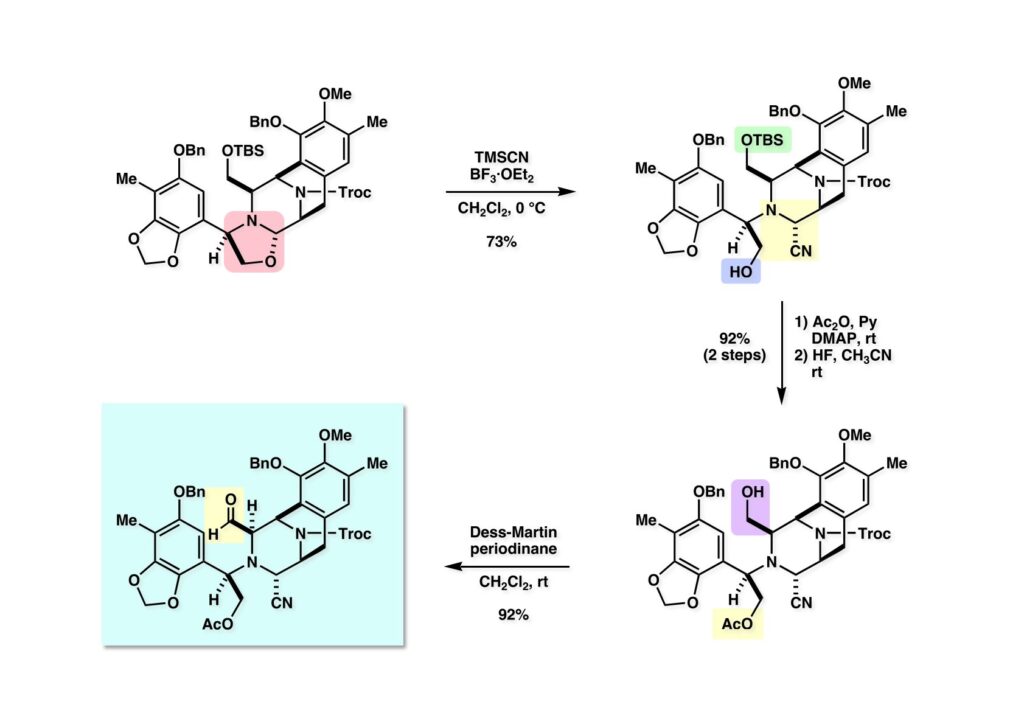
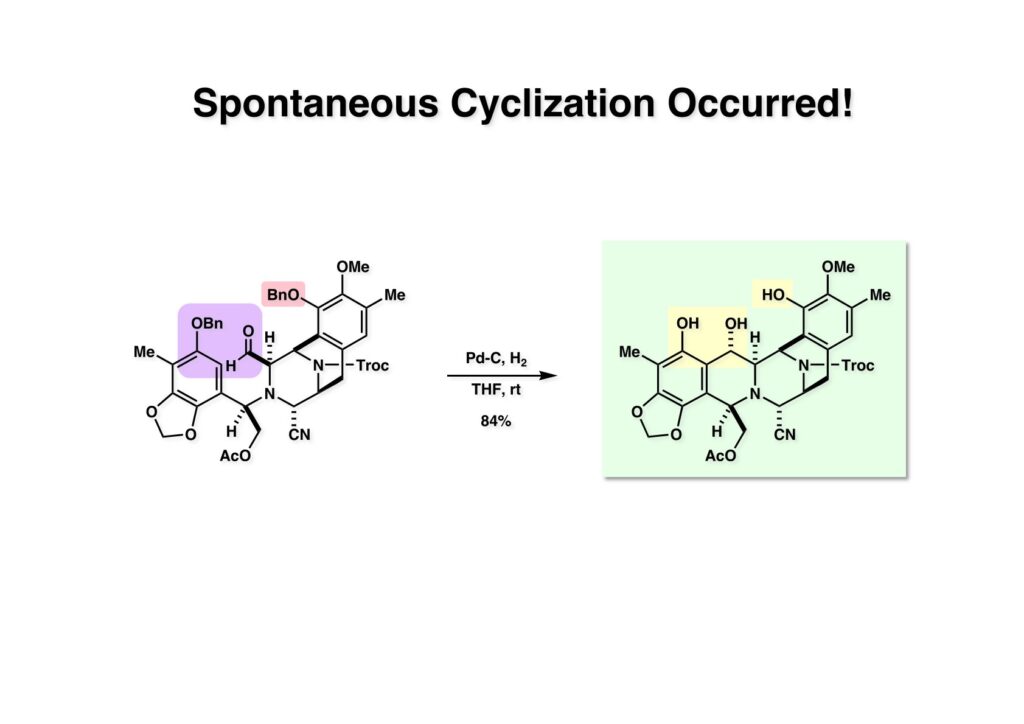
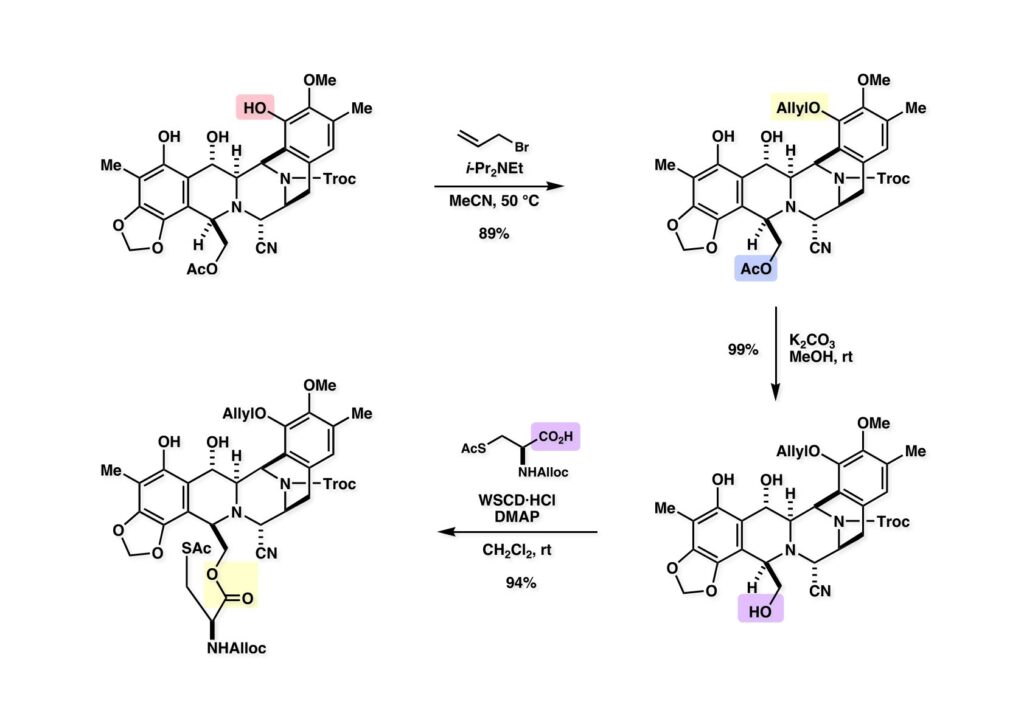
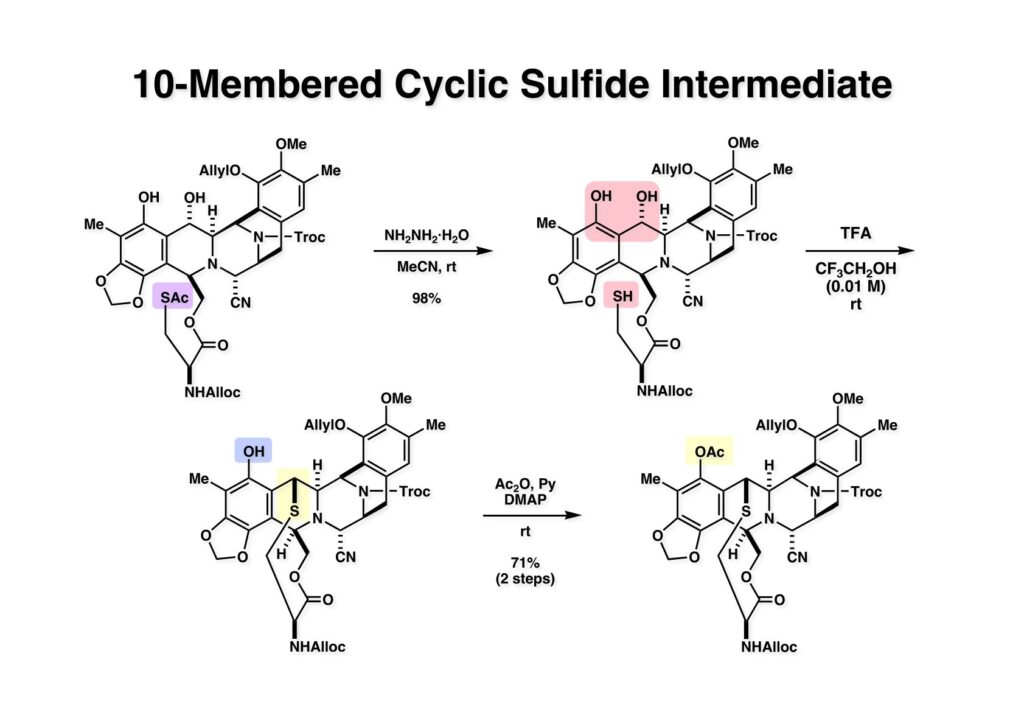
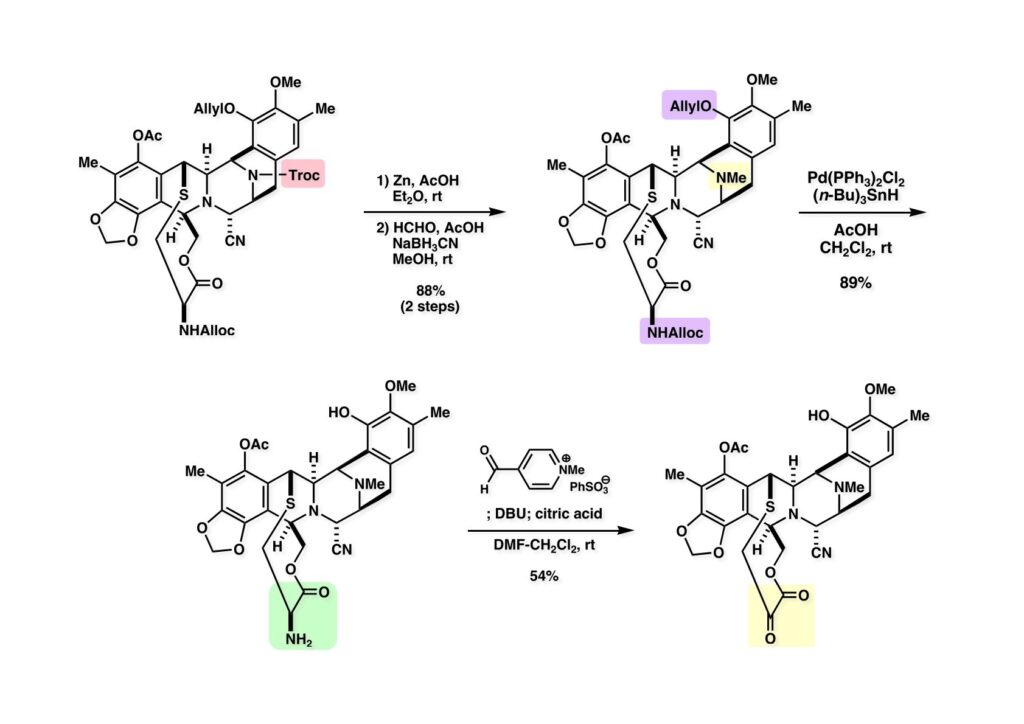
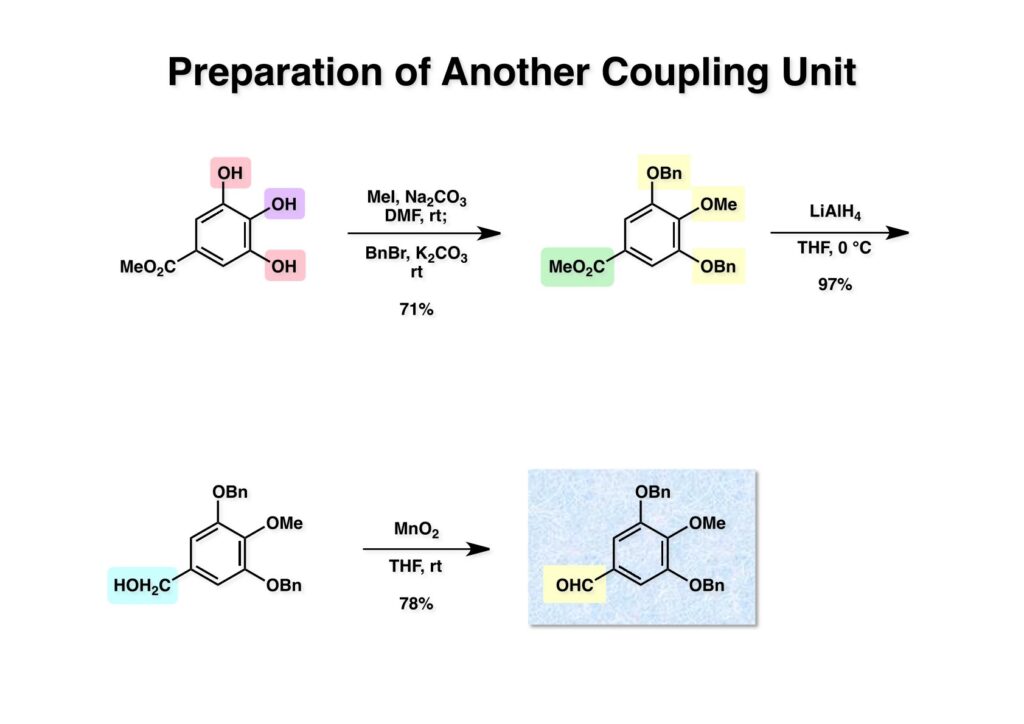
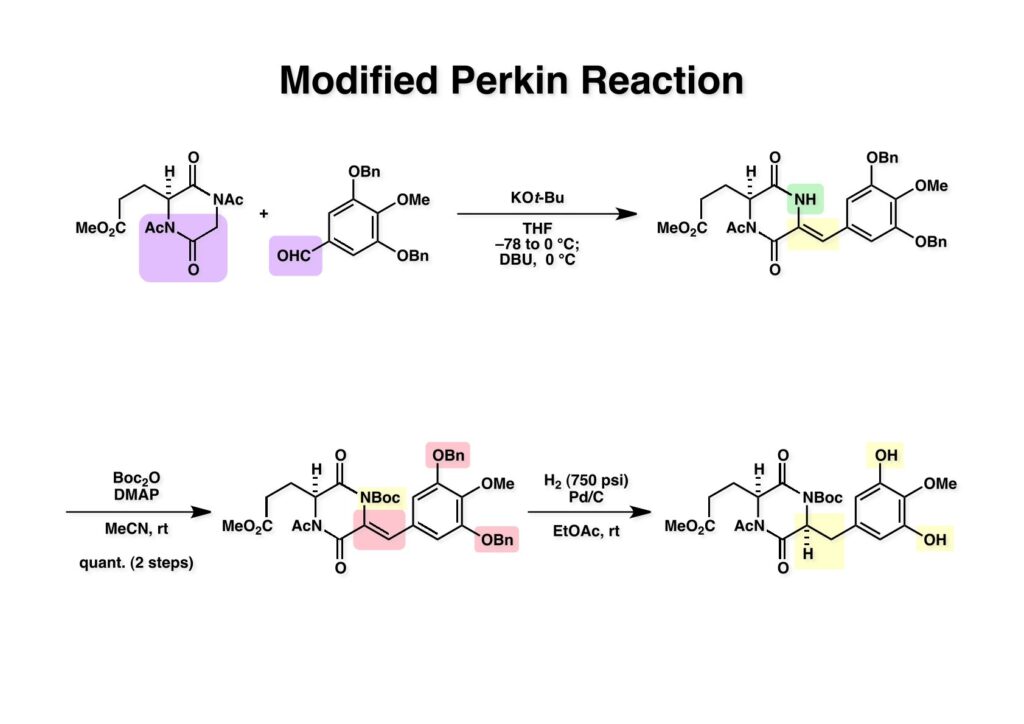
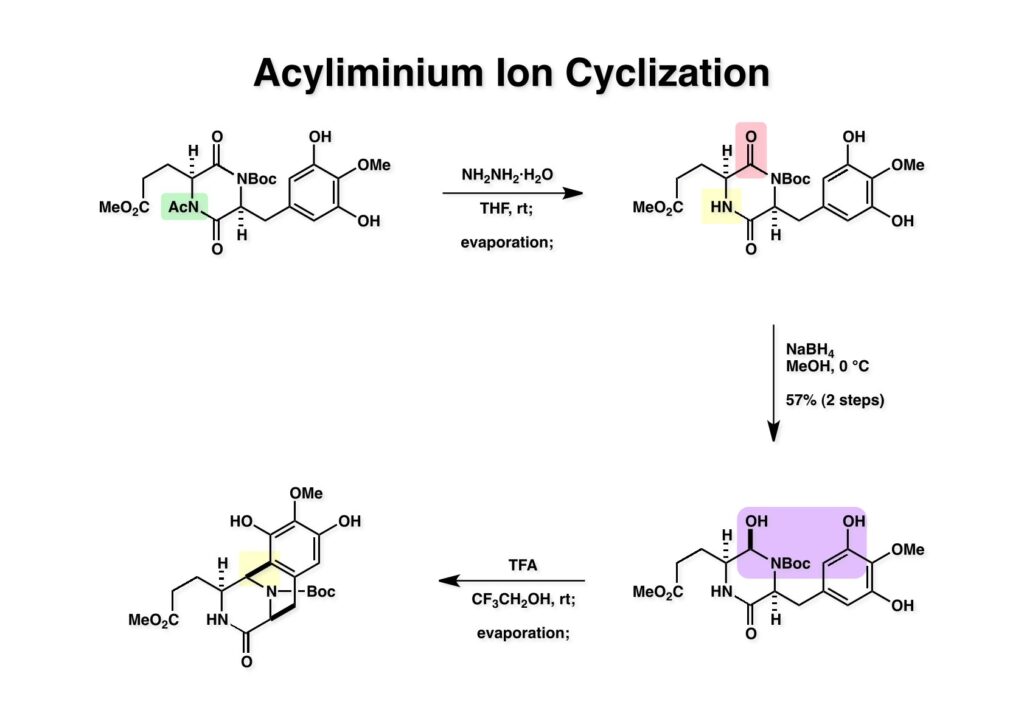
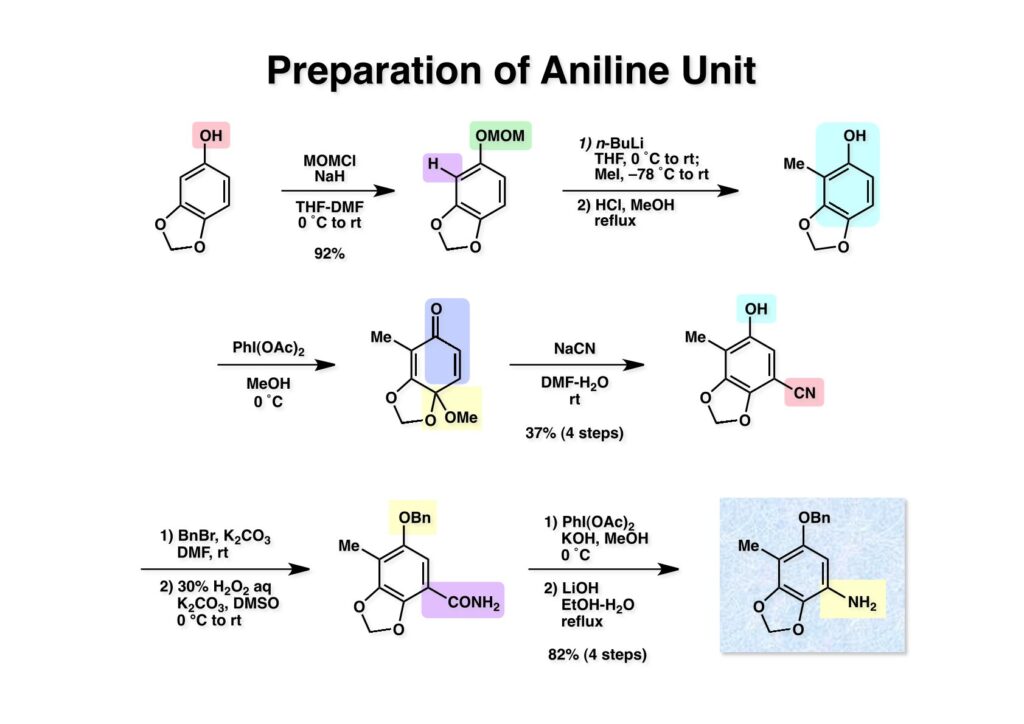
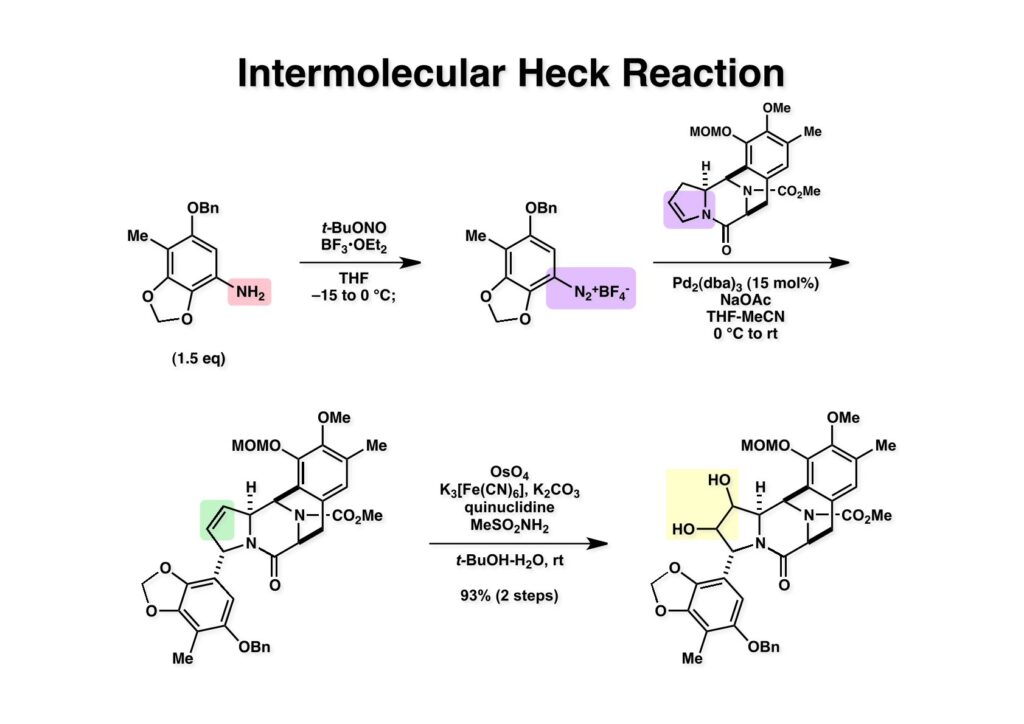
[a.k.a.]はalso known as 「又の名は」という言い表し方で、たまに見かけるが、ちょいと使ってみただけである。 E T-743は群体ホヤからは微量にしか得られないのでPharmaMarでは発酵産物であるcyanosafracin B (1-1) からET-743を合成している。詳しくは文献を参照されたい。 ET-743の最初の全合成はハーバード大学のCorey教授が達成したが、ポスドクのDavid Ginでなければ完成しなかっただろうと思う。Davidとは米国で何度も会ったことがあり、イリノイ大学からニューヨークのSloan Kettering Cancer Centerに移る前にも相談された仲だった。非常に独創的で有能な有機化学者だったが44歳で急逝したのは残念の極みだった。おそらく無理して働き過ぎたのが一因の心臓麻痺だと思うが、体調が悪くなるとすぐにサボってしまう私だからまだ生き長らえているのかも知れない。 初期の頃の逆合成解析をここに示す。(1-1) のtetrahydroisoquinoline環の立体化学は右端の部分が画面に直角に突き出すように立っているのでPictet-Spengler反応で簡単に制御できることは自明だ。(1-3) のベンジル位の水酸基は酸性条件でquinone methideか安定カチオンを形成することが期待できるのでC-S結合の形成が可能である。(2-3) のようなジアルデヒドは片方がphnolic cyclization、もう片方はヘミアミナールを形成することが期待できる。このジアルデヒドは (2-2) の1,2-ジオールを開裂させれば良い。ということで、(2-2) のヒドロキシメチル基の立体化学の制御は棚に置いておいて、まず (2-1) のような化合物を合成するにはどうすれば良いかを次ページのように考えてみた。 (1-1) のアミノ基は (1-2) の水酸基の反転によるものとし、パラ位をブロックしたフェノール (1-3) によるPictet-Spengler反応で (1-2) を構築することにした。(1-3) のヘミアミナールを開館して水産基とアルデヒドでラクトール (2-2) を構築すると、D-グルコース (2-1) からの展開が見えてくる。 グルコース (1-1) から5段階でエポキサイド (1-2) に変換するルートはよく知られており、何も見なくても私は書けるが、読者は考えてみたら?このエポキサイドをTsNH2と炭酸セシウムで加熱すると開裂が起こり、生じた水酸基をメシル化して (1-3) を得た。アセトナイド (1-3) を加メタノール分解すると (2-4) のアノマー混合物になるが、続いてSnCl4で処理するとβ-体に収束する。ここでNaOHを用いるとN -トシルアジリジンが生成し水酸基をシリル化して (2-3) が得られた。この活性化されたアジリジン (2-3) はCuI存在下でGrignard試薬 (2-2) と反応させると容易に開環して (2-1) を与えた。 次に (1-1) のトシルアミドのNHをBoc基で保護してからTBS基をTBAFで外してアルコール (1-2) を得た。この水酸基をTriflateに変換した後にLiN3と加熱してアジド体 (1-3) が得られた。フェノールのMOM基はMe3SiBrを用いて除去し、次にTFA /CH2Cl2処理でBoc基を外して (2-2) が得られた。ここでフェノールのパラ位をブロックするためにPyHBr3でブロモ化し (2-1) に変換した。フェノールは一般的にパラ位の方がオルト位より反応性が高い。 (1-1) を含水トリフロロ酢酸と加熱するとアシルイミニウムイオンを介した環化が進行して目的とするビシクロ体 (2-2) が高収率で得られた。(2-2) はフェノールのメチル化、ベンジル基のBCl3による除去、そしてRh/Cを触媒とするアジドの接触還元で (2-1) に変換された。ここではBr基は残す必要もなかったがロジウム系の触媒はsp2炭素に結合したハロゲンを残したまま二重結合などを還元できる。 次の課題は (1-1) から (1-2) への変換で、HOCH2基の立体化学を制御しながら左端の芳香環を導入しなければならない。アミノアルコールはcis 配置なので (2-1) を酸化してイミン (2-2) を作れば、電子豊富なAr基をβ面から付加させて目的物 (2-4) が合成できる。 (1-1) のアミノアルコールに活性エステルを持つphenyl bromoacetateを用い、アミンのアルキル化後に直ちにラクトン環が構築できるようにした。但し、6員環ラクトンは不安定で開環しやすいのでアミン塩基を使うことは避けた。代わりに使ったのはプロピレンオキサイドでHBrが生成すれば直ちに反応してブロモヒドリン(中性)となる。この方法はアミノ酸の塩酸塩などを中性条件でフリーにするのに使われたらしく、その為に使った事はないが知識としては持っていた。アミン (1-2) を四酢酸鉛で酸化するとイミン体 (2-1) が得られた。これをTFA存在下でフェノール (2-2) と反応させると予想どおりβ面から付加が進行して目的物 (2-3) が高収率で得られた。 (1-1) までは順調に進んできたが、アミンの保護に手こずっている最中に、「安価なグルコースを出発物に使ったもののけっこう長ったらしいルートになってきたなー」と、少々嫌気がさしてきた。おそらくET-743の全合成はこのままプッシュしていけば完成するだろうけど、これでは実用的な合成とは程遠くなる…。こういう悩みが頭に浮かんでくると、もっと良いルートはないものか、と考え始めるのがいつもの事で、浮気と言えば浮気なのだが、初志貫徹が必ずしも美しくないのもよくある事である。まあ、走りながら考えるって言うのが面白いところである。 溝呂木-Heck反応、と言ってもアメリカに居た時はHeck反応としか言ってなかったので、ここでもHeck反応と書いてしまうが、対象となるオレフィンは殆どが炭素置換の化合物で例外的にジヒドロピランが頭に入っているくらいだった。しかし、(1-1) のように窒素が2つも結合しているようなオレフィンにHeck反応が通用のかどうかは知らなかった。もしこの反応が成功して (1-2) に変換できれば今までとはずいぶん違った合成ルートになると思い、とりあえずモデル実験をやってみることにした。 ジケトピペラジン (1-1) は1985年に報告した我々の方法を用いて合成した(次ページ)。ジケトピペラジン (1-1) のアミドNHにBoc基を導入することでカルボニル基を活性化することが出来る。NaBH4還元して緩和な条件(CSAーキノリン)で脱水するとエナミド体 (1-2) が生成する。これをパラジウム触媒と反応させると定量的にビシクロ体 (2-1) が得られた。これはET-743の全合成に使えると思った瞬間である。 5 6 7 9 Tetrahedron Lett. , 26 , 2955-2958 (1985).ここからはHeck反応を鍵反応としたET-743の全合成について説明する。 (1-1) から (1-3) への逆合成解析は以前と変わりないが、アルドール型のphenolic cyclizationを期待して (2-3) に変換する。アミノニトリルはラクタムの還元とNaCN処理によって構築することとし、肝心のアルデヒドは何とかオレフィン (2-2) から導きたいと思った。(2-2) は (2-1) のHeck反応を適用すれば良い。 このエナミド体 (1-1) は (1-2) の脱水によって構築できるが、(1-2) は (2-1) からのAルート、もしくは (2-2) からのBルートが考えられる。 Aルートは (1-1) のアミド化か (3-1) の立体障害のあるアミンのアシル化を経由しなければならないので少々ハードルが高いと思った。特に後者はアミノ酸のラセミ化が心配である。 Bルートについても (1-1) の合成には困難さが付きまとうと思った。ところが (3-1) の構造をよくよく見てみると、N -acylamino acid amideということで、これはUgiの 4-component condensation (4CC) 反応が使えそうだということに気が付いた。 中間体 (1-1) をUgiの4成分連結反応で構築するには、アミン (1-2)、アルデヒド (1-3)、イソニトリル (1-4)、そしてカルボン酸 (1-5) を用いれば良い。これをメタノール溶媒中で加温すれば出来上がりという具合だ。 アミノ酸 (1-1) はデヒドロアミノ酸 (1-2) の不斉還元で合成することとし、(1-2) はアルデヒド (1-3) から容易に得られることは分かっている。一方、アミン体 (2-1) のメチル基はフェノール (2-2) から変換することにし、以前に確立した (2-4) とフェノール (2-3) からのアミノ酸合成法を使えば (2-2) が得られると考えた。 まず、右端部分のアミノ酸 (1-2) の合成について説明する。 市販の3-methylcatecholを注意深くトシル化すると空いている1位のフェノールだけが反応して (1-2) が得られる。これを臭素でブロモ化するとパラ位が反応して (1-3) が生成する。次にフェノールのメチル化、トシル基の加水分解、得られたフェノールのMOM化で (2-1) が高収率で得られた。(2-1) を低温下でn -BuLiによるハロゲンーリチウム交換の後にDMFを加えるとアルデヒド (2-2) が得られる。そしてメタノール中でオルトギ酸メチルとCSAを使ってアセタール化することで (2-3) を得た。オルトギ酸メチルは系内の水と反応してギ酸メチルとメタノールになるので効率的な脱水剤と言える。 MOM基単独でもフェノールのオルト位はBuLiで脱プロトン化できるが、(1-1) の場合はジメチルアセタールがあるのでより容易にリチオ化でき、ヨウ素を加えることでヨウ化体 (1-2) が得られる。次に塩酸でMOM基とアセタールを除去し、フェノールのベンジル化により (2-1) を得た。このアルデヒドを文献に従いHorner-Emmons反応でデヒドロアミノ酸 (2-2) に変換した。 デヒドロアミノ酸エステル (1-1) の不斉還元はMark Burkが報告したDuPhosをリガンドとしたロジウム触媒 (2-2) を用い、(1-2) がほぼ定量的に94% eeで得られた。彼はDuke大学の准教授の時に忽然と姿を消したが、面白い噂話をアメリカで聞いたことがある。(1-2) を加水分解してN -Bocアミノ酸 (2-1) を得た。 さて次はアミン体 (1-2) の合成について説明する。中断した合成経路ではイミン (2-1) に対する電子豊富なフェノール (2-2) の付加反応によってヒドロキシメチル基の立体化学を制御した。従ってこれと同じ手法で光学活性なアミノアルコール (1-2) を合成しようと計画した。 市販の(R )-phenylglycinol (1-1) をプロピレンオキサイド存在下でphenyl bromoacetate (1-2) と反応させて (1-3) を得た。これを加温してラクトン (1-4) に閉環しようとしたが、不安定で低収率でしか得られなかった。γ-butyrolactone(5員環)は安定だが、δ-valerolactone(6員環)は不安定で安定剤入りのを冷蔵庫で保管しなければならない。(1-4) を (1-5) に変換しようとしたが、これも不安定で得られなかった。そこでエステルでもt -ブチルエステルは求核攻撃を受けにくいということで、(1-5) のメチレンの部分にメチル基を2つ導入することにした。市販の(R )-phenylglycine (2-1) をメチルエステルに変換してからアミノ基をBoc化して (2-2) を得た。これに過剰のMeMgBrを反応させると三級アルコール (2-3) が得られた。次にBoc基を塩酸で外してアミノアルコール (2-4) を得た。 アミノアルコール (1-1) を前回同様にphenyl bromoacetateと反応させて (1-2) を得た後にトルエン中で環流することでラクトン体 (1-3) を得た。これを四酢酸鉛で酸化してイミン体 (1-4) に変換した。相方のフェノール体 (2-4) は市販のセサモール (2-1) から容易に合成できる。まずフェノールをMOM化してからn -BuLiでオルト位をリチオ化し、(MeO)3Bを加えてボロン酸エステルにする、これを単離精製することなく過酸化水素で酸化するとフェノール (2-3) が高収率で得られた。(2-3) のイミン (1-4) への付加はTFAを使って-10度で行い、付加体 (3-1) が単一の生成物として得られた。 (1-1) のフェノール基をメチル基に変換するために、まずTf2OでトリフレートにしてからラクトンをNaBH4で還元してジオール (1-2) にした。3級水酸基存在下で1級水酸基をシリル化し、得られた (2-1) をMeZnClを用いた根岸カップリングの条件に付すことでメチル体 (2-2) が得られた。 1,2-アミノアルコールは1,2-ジオールと同じく四酢酸鉛でC-C結合を切ることが出来る。(1-1) も容易に反応が進行してイミン (1-2) が得られたが、それを単離することなくNH2OHと反応させることでアミン (2-1) が得られた。おそらく現在ではもっと短工程で (2-1) が得られるだろうが、比較的大量合成が可能なルートではあった。 ミュンヘン工科大学のIvar Ugi教授の4成分連結反応は素晴らしい反応だが、ヒントになったのはPasserini反応 p -methoxyphenyl isocyanideはp -アニシジンをクロロホルムとNaOHで加熱する方法で合成した(大量合成が容易で蒸留で精製できる)。イソニトリルを使った後はフラスコなど全てをドラフト中で希塩酸に浸してホルムアミドに水和してから洗浄しなければ実験室中が臭うから気をつけること。p -Anisylと記述したのは間違いで、正くはMPMと書くべきだった) TBAFを使って (1-1) のTBDPS基を外してからアセテートに変換し、TFAでBoc基とMOM基を外すと (1-2) が得られるが、これを単離することなく酢酸エチル中で還流するとジケトピペラジン (2-2) に閉環する。Boc基を外す時にアニソールを共存させるのはt -ブチルカチオンをトラップするためで、チオアニソールが使われることもある。次に (2-2) のフェノールをメシル基で保護して (2-1) を得た。 このページの反応経路は読者は十分に把握していると思う。まずラクタム (1-1) を活性化するためにN-Boc化して (1-2) にする。この方法はアミドやラクタムの加水分解や加アルコール分解が容易でないことからPaul Griecoが報告した。Boc基は嵩高いので攻撃されず、アミドのカルボニル基はイミドのそれとなるので求核攻撃を受けやすくなるという仕組みだ。(1-2) をNaBH4で部分還元すると (2-2) が得られる。少量の硫酸を加えるのが必須ではないが、反応加速と過還元を防ぐ役割が期待できる。次にCSA-キノリン存在下で加熱することにより脱水が進行してエナミド体 (2-1) が高収率で得られた。 モデル実験の時よりは些か収率が低かったが (1-1) のHeck反応は期待どおり進行してビシクロ[3.3.1]体 (1-2) が得られた。 ET-743は左の芳香環のフェノールがアセテートになっているので、ここでメシレート (1-1) を加水分解してアセテート (1-2) に変換したのだが、これが不安定で維持し続けられなかったのでタイミングが早過ぎたと反省している。それではベンジルエーテルにしておけば良かったか、と言えばそうでもない。大いなる計算違いだったのは (1-1) のオレフィンに対してハイドロボレーションが全く進行しなかったことだ。そこでオレフィンをエポキシ化しようとしたらベンジルエーテルでは芳香環が酸化されてしまい、やむ無くアセテートにしておいたという経緯がある。(1-2) のBoc基をTroc基に付け替えて、得られた (2-2) のオレフィンを如何にしてアルデヒド (2-1) に変換するかが大きな問題となった。 Hydroborationが不調だったので、何とか立体化学を制御しながらアルデヒドを構築するためにビシクロ[3.3.1]系のexo側からハイドライド攻撃しようと企画した。オレフィン (1-1) の酸化はDMDO (dimethydioxirane) のみが良好な結果を与えた、反応溶媒はアセトン-MeOHだったのでエポキサイド (1-2) の生成完了後にCSAを加えることでメトキシ体 (2-1) が得られた。次にTFA存在下で (2-1) のNaBH3CN還元を行うと、アシルイミニウムイオン (3-2) を経由して目的物 (3-1) が単一生成物として得られた。図には74%とあるが最終的には94%の収率に改善された。 (1-1) の水酸基をTBS化した後に2つのアセテートを加メタノール分解して (1-2) に変換したが、なぜguanidine nitrateを使う条件になったのかは記憶に無い。おそらくTroc基がCO2Me基に変換されるのを防ぐためだったと思うが。(1-2) のフェノールを常法でベンジル化して (2-2) を得た。通常ラクタムを還元してヘミアミナールで止めるのはトリッキーだが、(2-2) をRed-Alで還元すると、隣にHOCH2基があるのでoxazolidine環が形成されてそれ以上の還元が起きないため無事に (2-1) が得られた。 ヘミアミナールは非常に不安定なので (1-1) をTMSCNとBF3·Et2Oで処理して安定なアミノニトリル (1-2) に変換した。次に水酸基をアセチル化し、TBAFでTBS基を外して (2-2) を得た。この水酸基をDess-Martin酸化してアルデヒド (2-1) を得た。とにかく保護基を頻繁に変えるのが嫌だったが、取り敢えず目的地に向かって前進するのみだった。 (1-1) のベンジル基を加水素分解したところ環化体 (1-2) が直接得られてきた。これほど簡単にフェノール環化するとは思っていなかったけど…。 (1-1) には2つのフェノール水酸基が存在するが、おそらくベンジル位の水酸基との分子内水素結合により、右側のフェノールが選択的にアリル化されて (1-2) が得られた。ここでアセテートの加メタノール分解によりアルコール (2-3) に変換し、システイン誘導体 (2-2) と縮合してエステル (2-1) を得た。最終段階で先行研究 (Corey-Gin全合成)と同じ経路を辿るのは潔しとしないが、この場合は時間との戦いでやむ無く信念を曲げてCorey-Ginの軍門に降った😭。 (1-1) のチオアセテートをヒドラジンで注意深く外してチオール (1-2) にし、次にCF3CH2OH中でTFAを加えると、おそらくquinone methideを経由すると思われるが、大環状のスルフィド (2-1) が生成し、フェノールのアセチル化後に (2-2) が得られた。 ここで (1-1) のTroc基を亜鉛還元して落とし、還元的メチル化で (1-2) に変換した。次にフェノールのアリル基とAlloc基をパラジウム触媒とBu3SnHで外して (2-1) を得た。このアミンはHenry Rapoportが報告したbiomimeticな方法でケトン (2-2) に変換した。
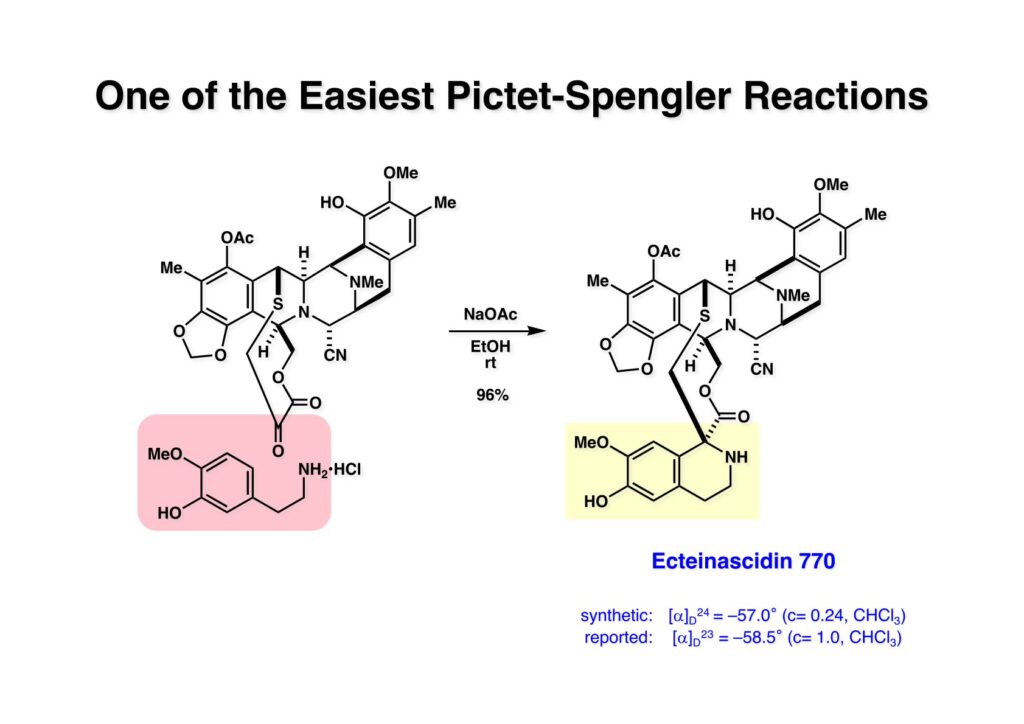
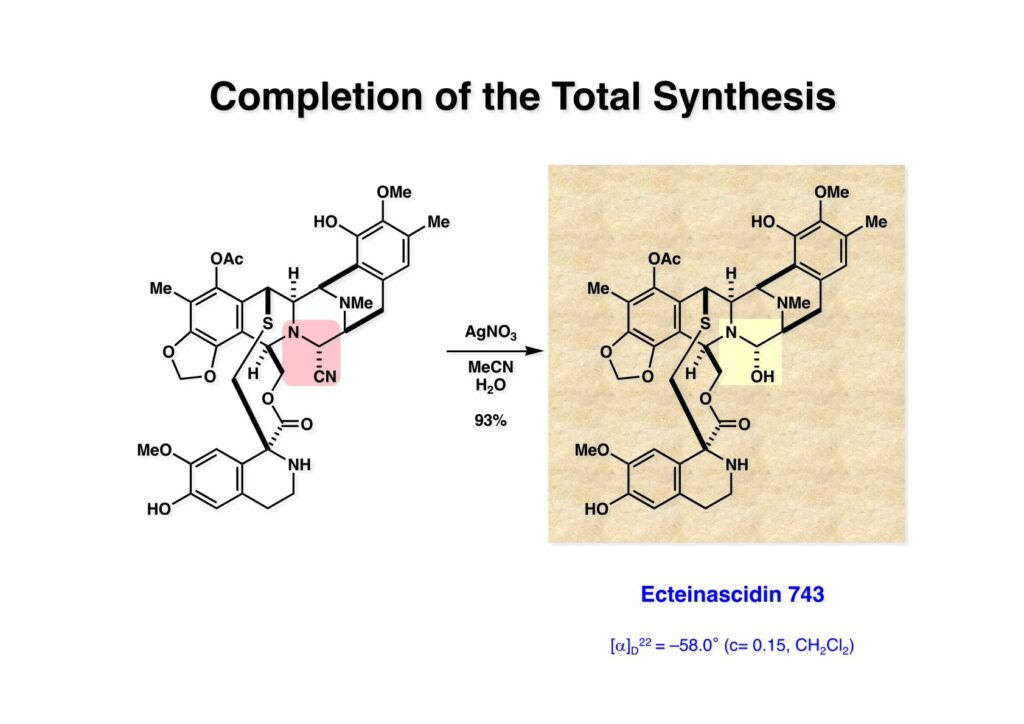
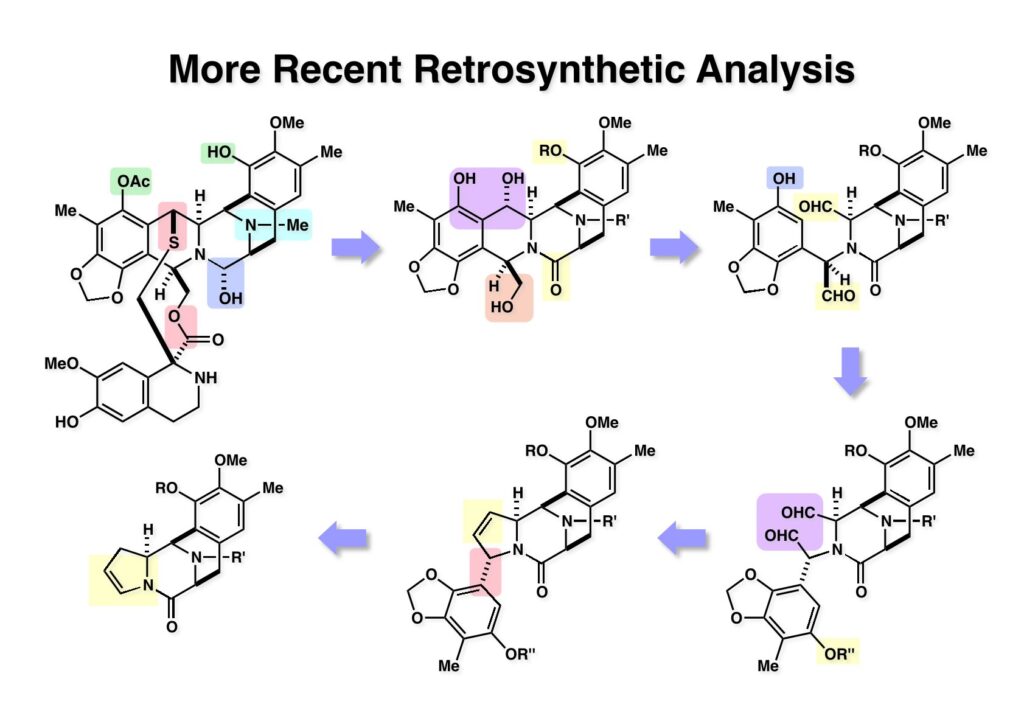
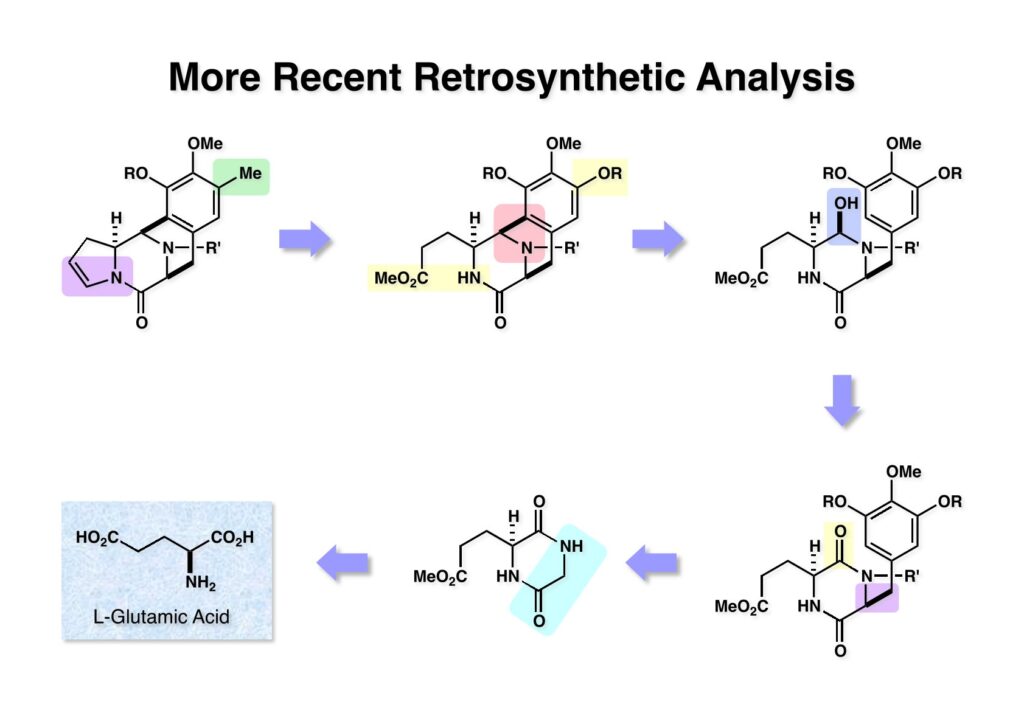
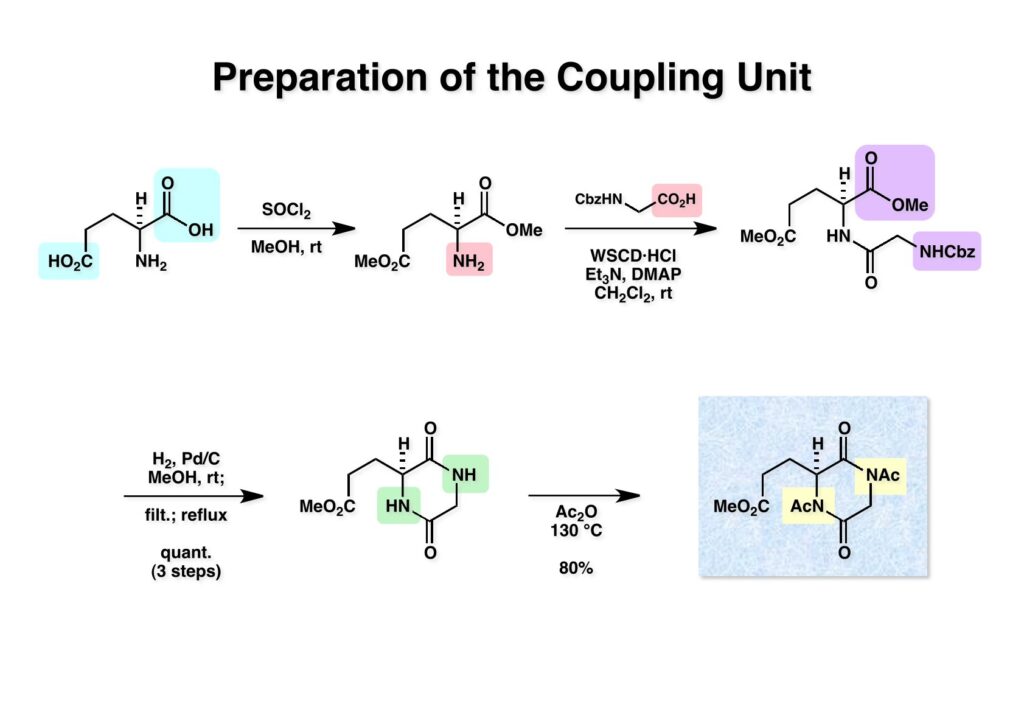
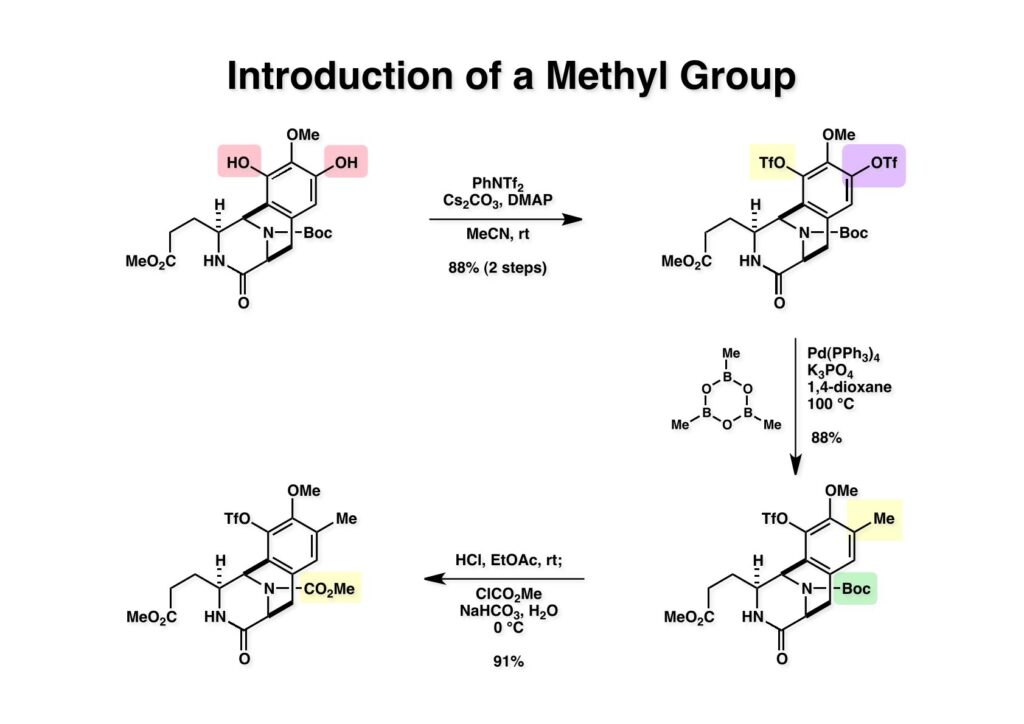
ケトン (1-1) とアミン (2-1) のPictet-Spengler反応は容易に進行し、ほぼ定量的にET-770 (1-2) を単一生成物として与えた。前にも話したがヘミアミナールは非常に不安定でアミノニトリルは安定である。アミノにトリルは塩酸で加熱するとアミノ酸になるが、含水アセトニトリル中で硝酸銀と反応させるとヘミアミナールに変換することができる。 ET-770 (1-1) をCH3CN-H2O中でAgNO3と反応させることにより目的とするET-743が得られた。以前、cyanocyclineをnaphthyridinomycinに変換したときに後処理でNaHCO3-NaClとCH2Cl2を用いて分液したところAgCNがAgClとNaCNになって直ちにcyanocyclineに戻ってしまったという苦い経験があったので、ここでもNaClは用いていない。 我々の第一世代全合成の問題点を挙げるとすれば、鍵反応となるUgiの4CC反応は良しとしても、得られた生成物 (1-2) の紫色でハイライトした保護基を後の工程で全て別の保護基に付け替えなければならなかったことだ。このために工程数が増えてしまった。もっと良い工夫をすれば多少は工程数を減らすことが出来たかもしれないが、第二世代の全合成はこの反省点に立って実行されなければならないと思った。 第二世代のET-743全合成の逆合成解析においての基本的な考え方はいくつかあるが、まずジヒドロピラン (DHP) のHeck反応は2位で起きて二重結合は3-4位へ移動することはよく知られている。ということは、(2-1) のようなエナミド体の場合も2位に反応が起きて、二重結合が移動した (2-2) が得られることが期待できる。立体化学の制御はビシクロ[3.3.1]系で芳香環が画面の上に直立している状態だから画面の下から反応が起きることは自明である。この二重結合を切断してジアルデヒドにすれば、第一世代合成でフェノール環化が室温でスムーズに進行したことを考えれば (1-3) から (1-2) への変換も容易であると思われる。ということで、如何にしてエナミド体 (2-1) を合成するかが鍵となる。 (1-1) のようなエナミド構造を構築するためには (1-2) のようなC3ユニットが必要で、このビシクロ[3.3.1]系化合物を構築するのは (1-3) から酸性条件でacyliminium ionを発生させれば良い。(1-3) のような構造は (2-2) のジケトピペラジンを用いれば合成できるので、市販のL-グルタミン酸 (2-1) を出発物とすることにした。 L-グルタミン酸 (1-1) をメタノール中塩酸で加熱してジメチルエステル (1-2) に変換した。HClタンクを使うのはレギュレーターの掃除などで面倒なので、それほど大きいスケールでない時にはSOCl2かMe3SiClを使って系中で塩酸を発生させる。次にアミンとN -Cbz-glycineとを縮合してアミド (1-3) を得た。Cbz基を水添で外して触媒濾過後に加熱するとジケトピペラジン (2-1) が得られる。これを無水酢酸中で130度に加熱するとジアセチル体 (2-2) に変換できる。 没食子酸メチル (1-1, methyl gallate) をMeI-Na2CO3で注意深くメチル化すると真ん中のフェノールだけが反応する。これはパラ位にエステルがあって電子を吸引しているために酸性度が上がっているためだ。次に未反応のフェノール水酸基をベンジル化して (1-2) を得た。(1-2) のエステルをLAH還元してアルコール (2-1) を得てから二酸化マンガンで酸化するとアルデヒド (2-2) が得られた。 ジアセチルジケトピペラジン (1-1) とアルデヒド (1-2) の混合物にt -BuOKを加えると縮合反応が進行するが、反応を完結させるために、より安全なDBUを加えて (1-3) を得た。(1-3) のラクタムを活性化するためBoc2O-DMAPと室温で反応させてBoc体 (2-1) に変換した。次は酢酸エチル中でPd/Cを触媒として高圧下で水添すると脱ベンジル化されたcis -ジケトピペラジン (2-2) が得られた。 (1-1) のアセチル基をヒドラジンで注意深く外し、得られた (1-2) のイミドカルボニル基をNaBH4で還元すると (2-2) が得られる。以前、これよりも単純な系で側鎖がプロピオン酸ユニットでなくメチル基の時にギ酸処理で環化しようとしたらメチル基が完全にエピメリ化を起こしてしまった。ところが CF3CH2OHを溶媒としてTFAでアシルイミニウムイオン環化をしたところ側鎖の立体化学を保ったまま環化した (2-1) が得られた。 ビシクロ[3.3.1]系化合物 (1-1) の構築が終わり、次は2つのフェノール水酸基をトリフレートに変換して (1-2) を得た。これをパラジウム触媒とtrimethylboroxine (2-1) を用いてSuzuki-Miyauraカップリングで立体障害の少ない方のトリフレートを選択的にメチル基にした (3-2)。次に塩酸を使って (3-2) のBoc基を外してClCO2Me-NaHCO3でメチルカーバメート (3-1) にした。何でBoc基の代わりにMeO2C基を始めから入れておかなかったかだって?それはアミドのNHにCO2Me基を入れるためにはLDAやNaHなどの強塩基を使う必要があり反応性の高いN -アセチルアミドが耐えられない可能性があるためだ。Boc基はBoc2OとDMAPを使えば室温でアミドをアシル化することができるので信頼できる。 (1-1) のメチルエステルをL-Selectrideで還元すると、通常はアルコールまで還元されるが、この場合はラクタムのNHがあるのでアルデヒドの段階で捕捉されて (1-2) が得られる。(1-2) の脱水にはCSA存在下にトルエン中で環流する条件を用いたが (2-2) の収率は期待するほど高くなかった。この条件では二量化が低収率の原因の一つで、様々な条件を試したが再現性よく収率を上げることが出来なかった。しかし、もっと良い条件は必ずあると今でも思っている。不要な (2-2) のトリフレートを加水分解し、フェノールをMOM化して (2-1) を得た。 Heck反応の相手である (3-2) の合成も意外に手間取って、これも改良すべき反省点であり絶対により良い方法があると思う。n -BuLiでリチオ化してからMeIを加えるとメチル基が導入され、次に酸性条件でMOM基を外すとフェノール (1-3) が得られる。しかし問題はこのフェノールのメタ位に簡単に窒素を入れる方法が無いことなのだ。(1-2) でメチル基の代わりに水酸基を導入したものとphenyldiazonium ionと反応させてアゾベンゼンに変換し、還元的にN-N結合を切ってパラ位にNH2基を構築したこともあったが、OH基をメチル基に変換しなければならなく、面倒なので断念したこともある。とにかく苦し紛れに (1-3) をメタノール中でDAIB [PhI(OAc)2]で酸化してジエノン (2-1) を得た。これをMe3SiN3-Me3SiOTfで処理してアジドを入れようとしたがうまく行かなかった。仕方ないのでNaCNを共役付加させた後に脱メタノール化された (2-2) を経由してアミノ基を構築することにした。(2-2) のフェノールをベンジル化して常法にしたがってアルカリ性過酸化水素で処理してニトリルをアミド (3-1) に変換した。次にメタノール中でDAIBを使った穏和な条件でのHofmann転位を実行してメチルカーバメートにし、さらにアルカリ加水分解をすることでアミン体 (3-2) を得た。 前ページで合成したアミン (1-1) とt -BuONOのTHF溶液にBF3·Et2Oを加えるとジアゾニウム塩 (1-2) が生成する。次にCH3CNとの混合溶液にしてエナミド (1-3) と0度から室温の間でHeck反応を行うとで目的物 (2-1) が得られたが、副生物との分離が困難であったためオレフィンのジオール化後に高収率で (2-2) を単離した。 ジオール (1-1) の過ヨウ素酸酸化はジアルデヒド体ではなくビスヘミアセタール体が生成してしまうのでNMRでもアルデヒドのピークは見えない。ここで多少のエピメリ化が起きていたがメタノールから結晶化することで単一物87%の収率で得られた。次にフェノールのベンジル基を加水素分解で除去して (1-2) として単離した。この化合物のフェノール環化は条件検討に時間を要したが、最終的にはm -キシレン中で120度に加熱することによりアルデヒドのエピメリ化もなく環化体 (2-1) が得られた。この溶液を-42度に冷却し過剰のRed-Alを加えて60度にまで加熱することでアルデヒドの還元、ラクタムの還元によるオキサゾロジン環の形成、メチルカーバメートのメチル基への還元が順番に進行して (2-2) が76%の収率で得られた。このページのデータは条件を改良する前のものである。(Macでは古いパワーポイントファイルのChemDrawが変更できなくて0から直さないといけないので失礼する) (1-1) を酢酸中でKCNと反応させるとオキサゾリジンが開環してアミノニトリル (1-2) が得られる。ここからはCorey-Gin合成を踏襲した経路でET-743を合成しているので詳しい説明は省略するが、システイン誘導体 (1-3) とWSCDで縮合し、アセチル基をヒドラジンで外すとチオール (2-1) が得られる。これをCF3CH2OH中でTFAを加えるとマクロ環化が進行する。次にフェノールをアセチル化して (2-2) を得た。 次いでAlloc基を外したアミンをRapoportの方法でケトン (1-2) に変換し、アミン (2-1) とPictet-Spengler反応、MOM基のTFAによる除去、そしてAgNO3-MeCN-H2O処理によるアミノニトリルからヘミアミナールへの変換によってET-743 (2-2) の全合成が完了した。まだまだ改良したい点は多々あったが、実用的合成には至っていない。