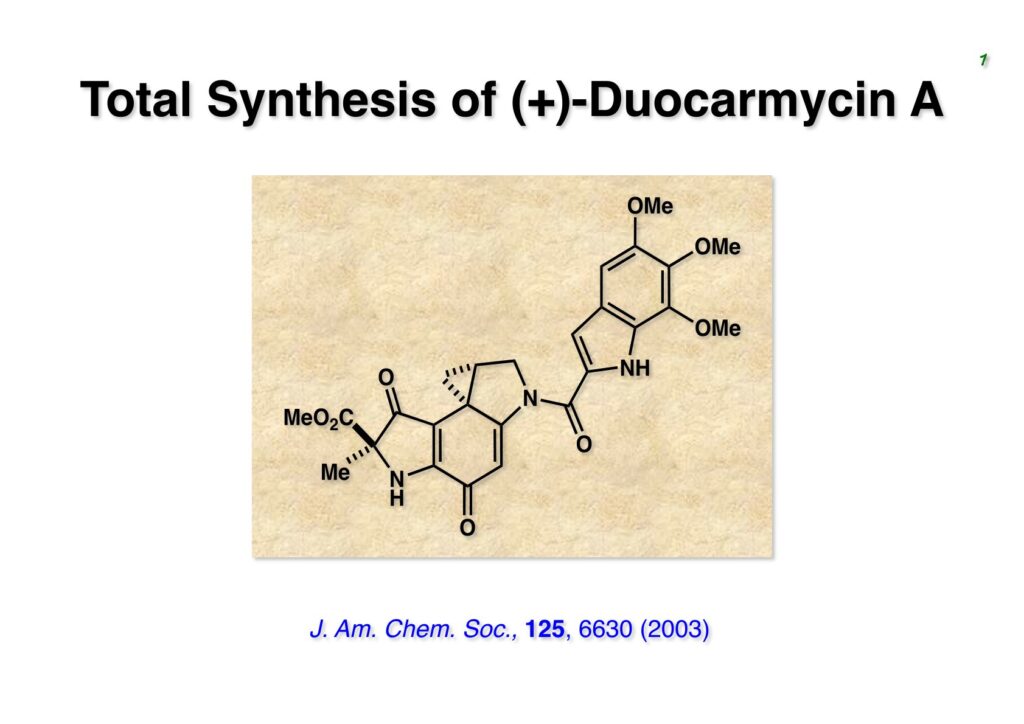
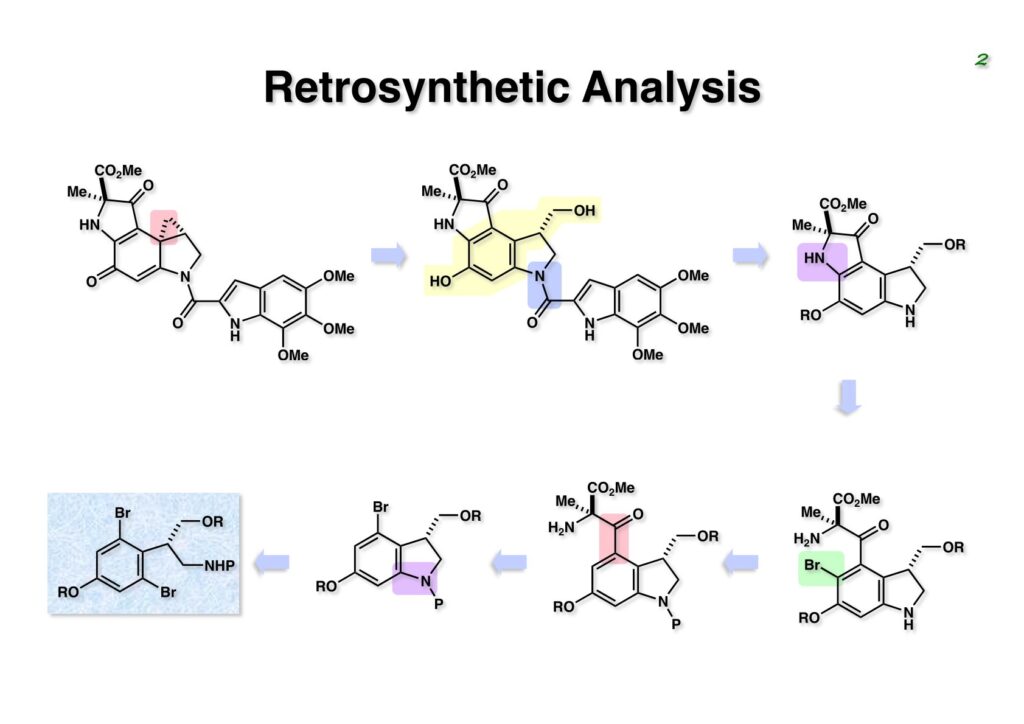
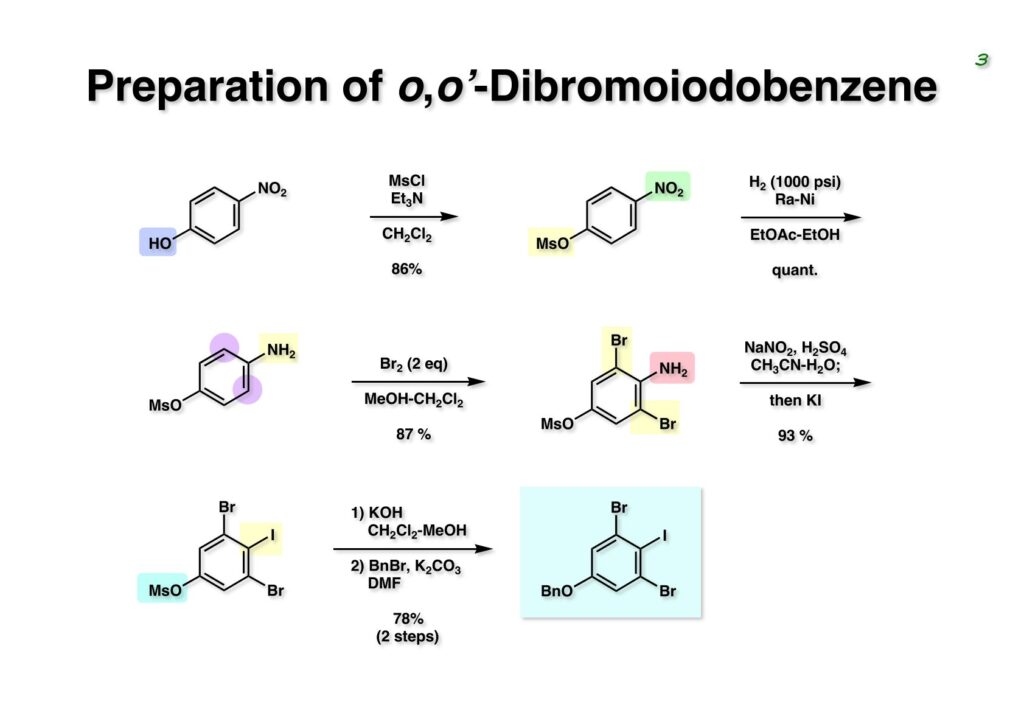
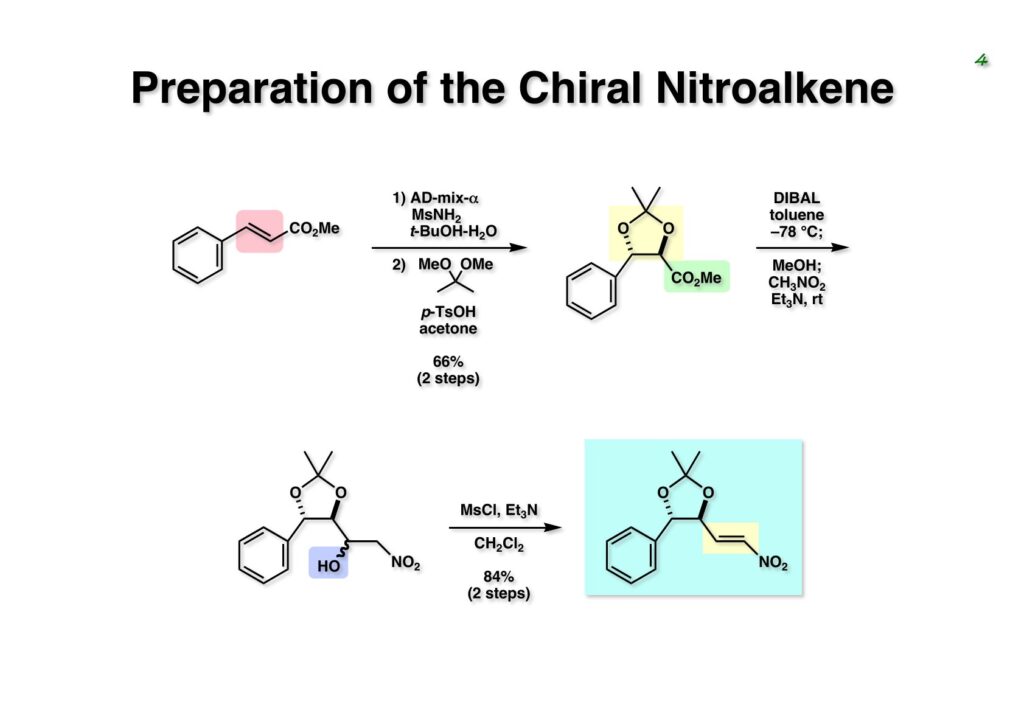
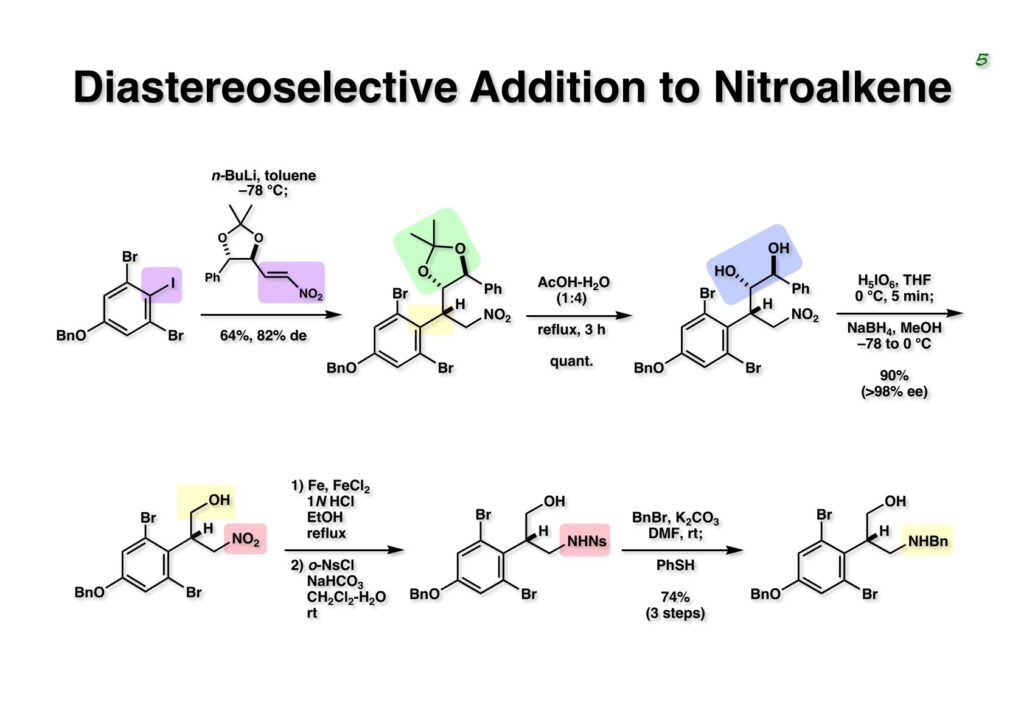
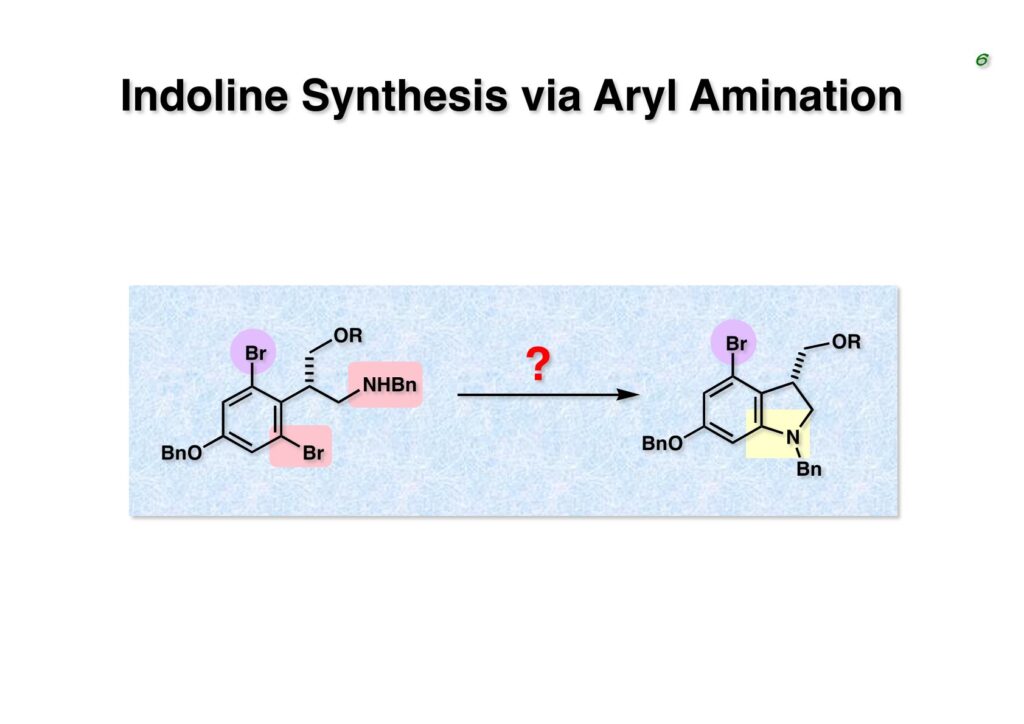
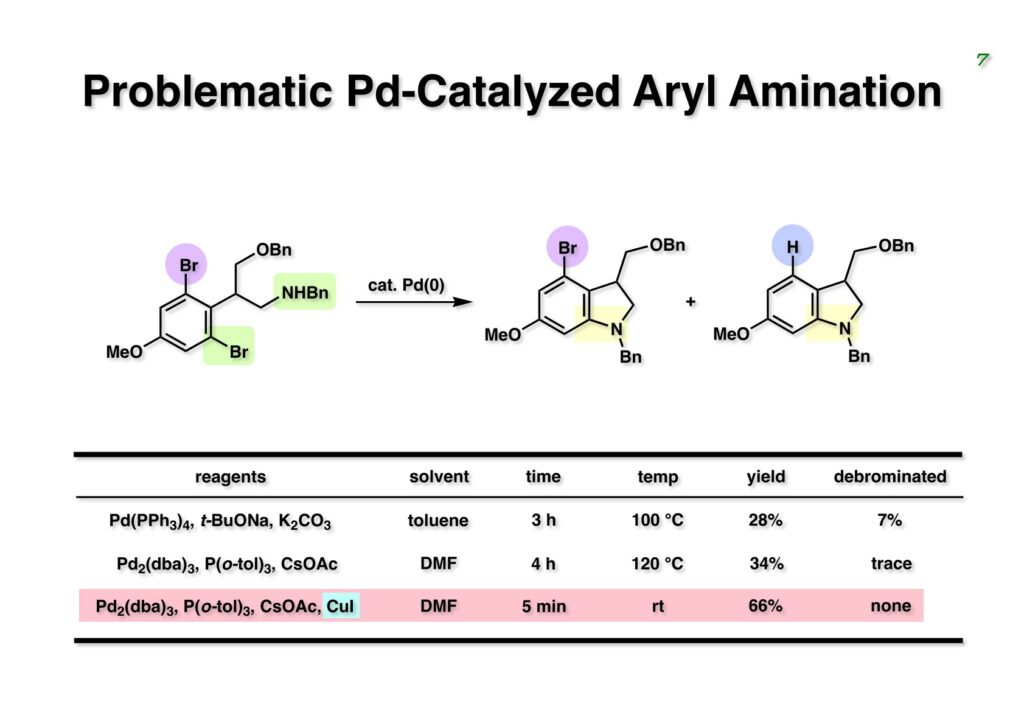
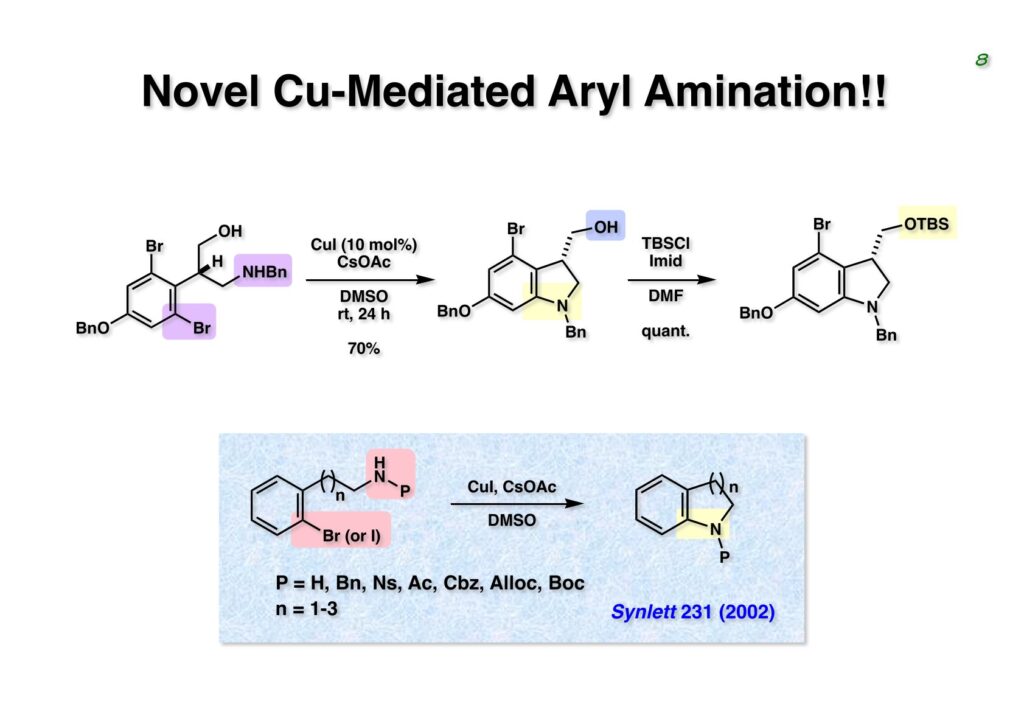
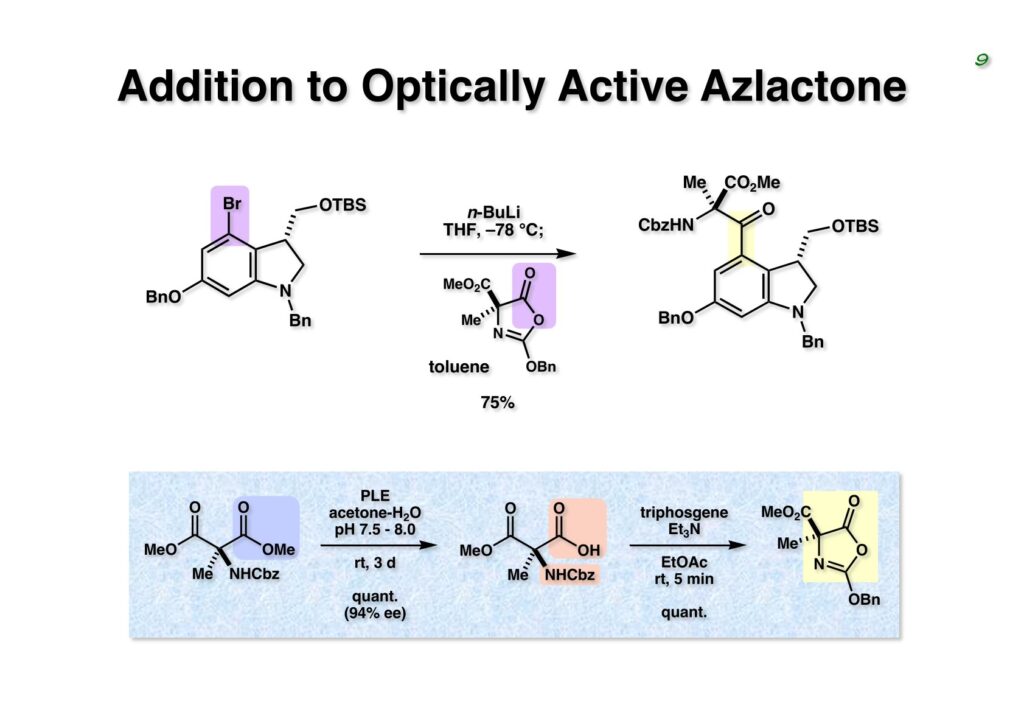
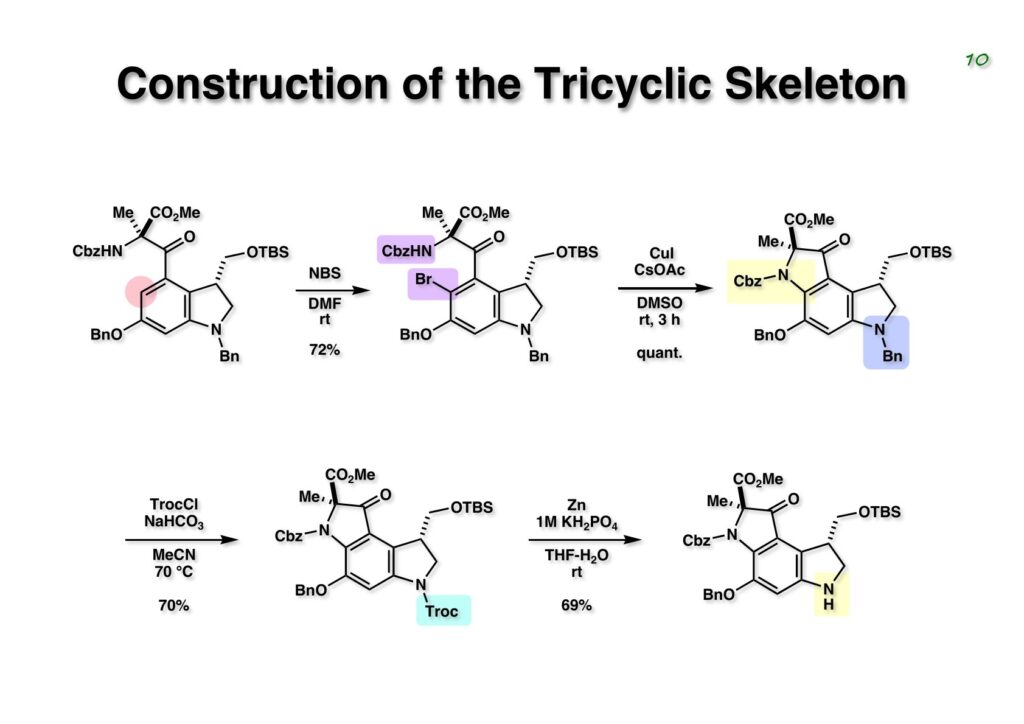
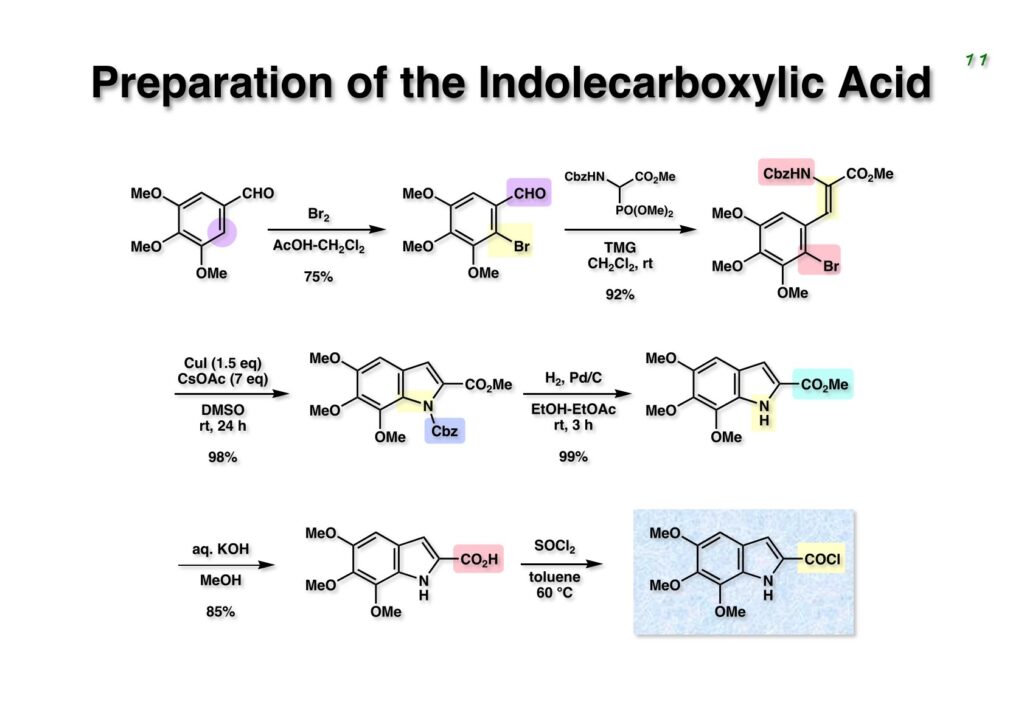
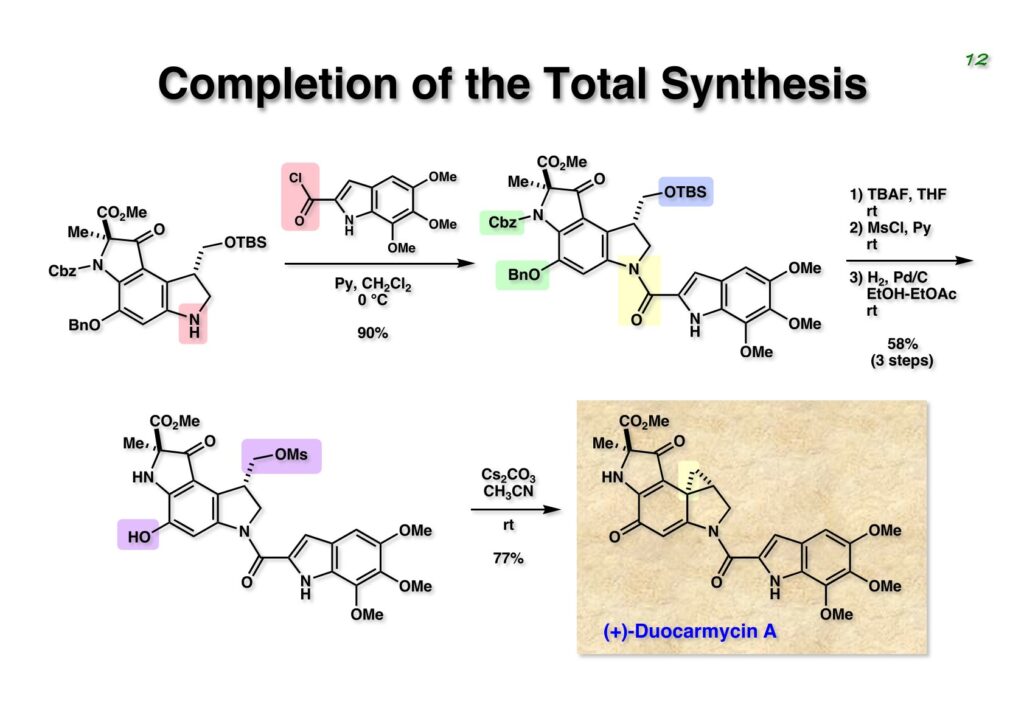
Duocarmycin Aは協和発酵で単離構造決定された抗腫瘍活性を持つユニークな天然物で、協和発酵のコンサルタントを1987年からやっていた私は、UpjohnのCC-1065 (東北大薬の徳山さんが全合成している)に似ていることもあって大いに興味を持っていた(私はUpjohnのコンサルタントもやっていた)。そこで、外国生活が長く、UCLAから博士課程に入学してきた山田健君に全合成をやってもらうことにした。理学部の奈良坂研から移ってきた黒川利樹君(現エーザイ)が開発した2,6-dibromoaryllithiumのニトロオレフィンへの付加反応を利用しているので彼の名前が論文に載っている。健ちゃん(私の次男が同名なのでいつもそう呼んでいた)のお父さんがトヨタに勤めていて、ドイツに転勤した際に一緒に渡欧し、確かベルギーの高校から米国のブラウン大学に行って学部研究はKathlyn Parker教授の下で行っている。日本の大学院に行きたいと岸先生に相談してから私の方にお鉢が回ってきたのだが、外国で高等教育を受けた人物が東大の修士課程に入るのは大変だから米国で修士号を取ってから博士課程の試験を受けるように助言した。その後、博士課程の入学試験は2番は何処にいるのか?というくらい高い点数で入ってきたのが記憶に残っている。このプロジェクトではCuI-CsOAcという条件で芳香環のアミネーションが高効率的に進行することが見つかった思い出深い研究である。健ちゃんは現在Novartisの研究員として米国Cambridgeで頑張っている。
“Total Synthesis of the Duocarmycins,” K. Yamada, T. Kurokawa, H. Tokuyama, and T. Fukuyama, J. Am. Chem. Soc., 125, 6630 (2003).