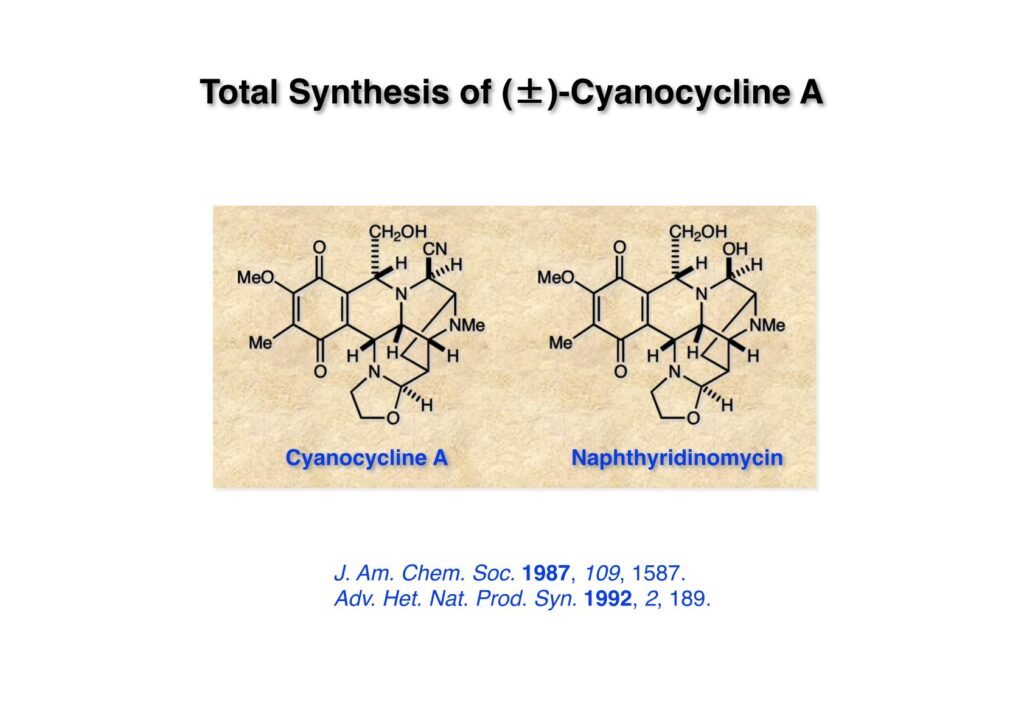
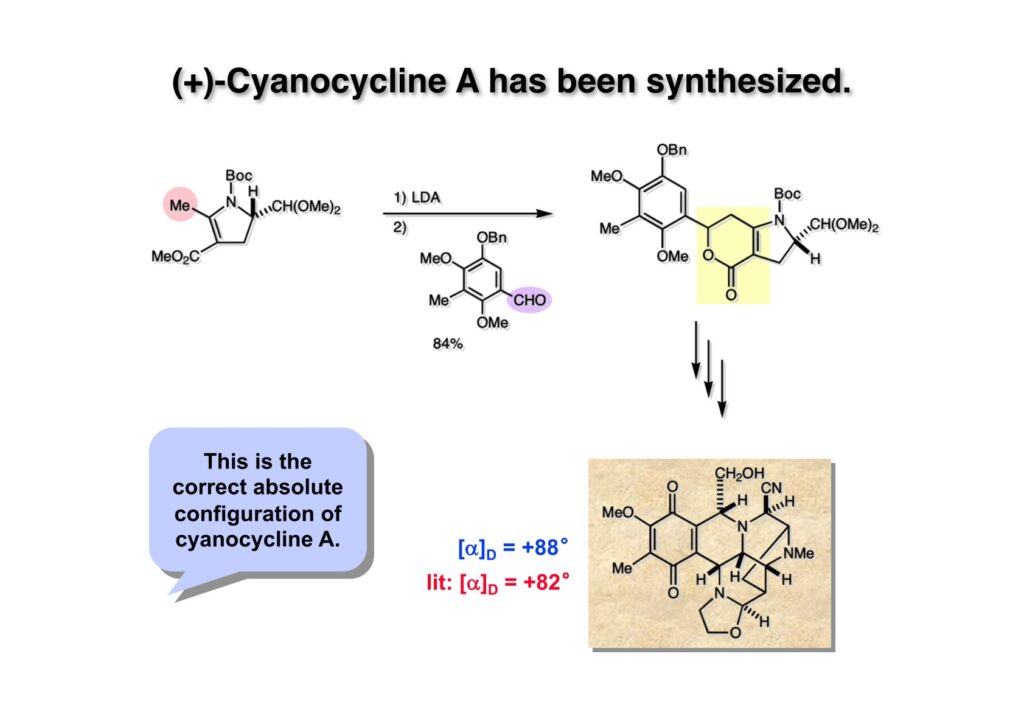
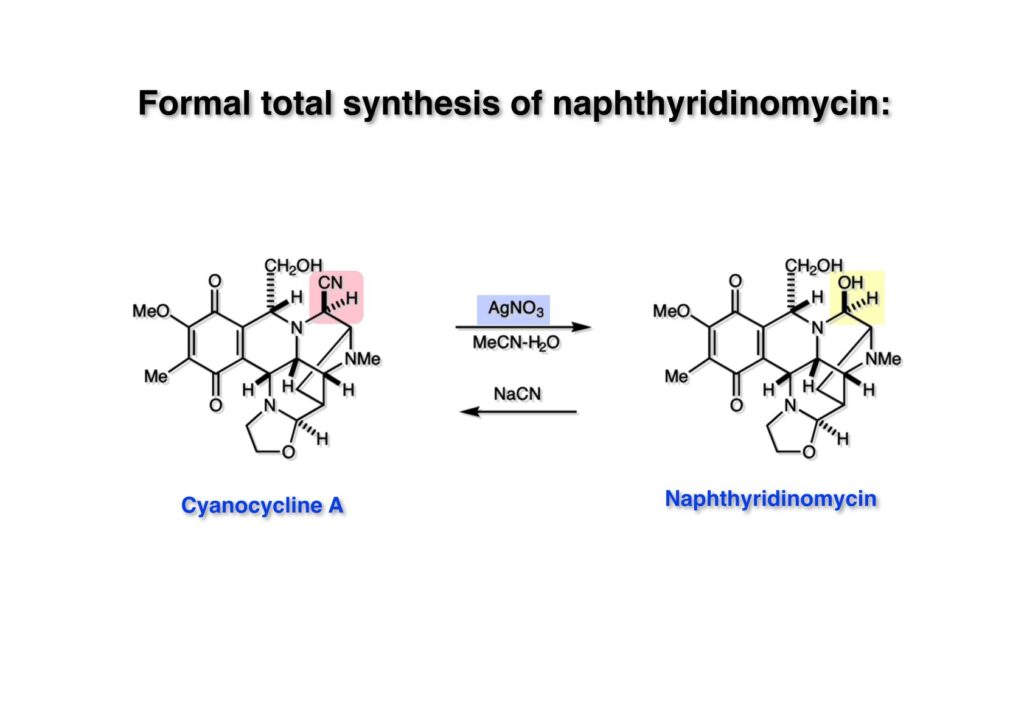
Cyanocycline Aは中外製薬の化学者が1982年に単離構造決定が報告された化合物であるが、当初の全合成の目的化合物ではない。私たちはnaphthyridinomycinの全合成をantibiotic 593Aの全合成完了後に目指していた。カナダのAyerst Laboratoriesが単離した化合物でモントリオール大学のHanessian教授のところでX線により構造解析がなされて1972年に報告された。Hanessianによると、非常に不安定な化合物で結晶を冷凍庫で保存していたところ全部分解してしまったそうだ。と言うことで、より安定なcyanonaphthyridinomycinすなわちcyanocycline Aの全合成で妥協したのだが、その後、光学活性なcyanocycline Aを合成して、中外製薬がX線解析の解釈を間違えて鏡像異性体の構造を提示していたことが判明した。そのついでにnaphthyridinomycinの全合成も完成したが、面倒なのでジャーナルには報告せず、Will Pearsonに依頼されたシリーズものの単行本のチャプターとしてまとめてお茶を濁してしまった。まあ、似たような論文を報告したくなかったと言うのが本当のところではあるけれど…。
“Stereocontrolled Total Synthesis of dl -Cyanocycline A,” T. Fukuyama, L.-P. Li, A. A. Laird, and R. K. Frank, J. Am. Chem. Soc. , 109 , 1587 (1987).
“Synthesis of Naphthyridinomycin,” in “Advances in Heterocyclic Natural Products Synthesis,” T. Fukuyama, Ed. by W. H. Pearson, JAI Press, 1992, pp 189-249.
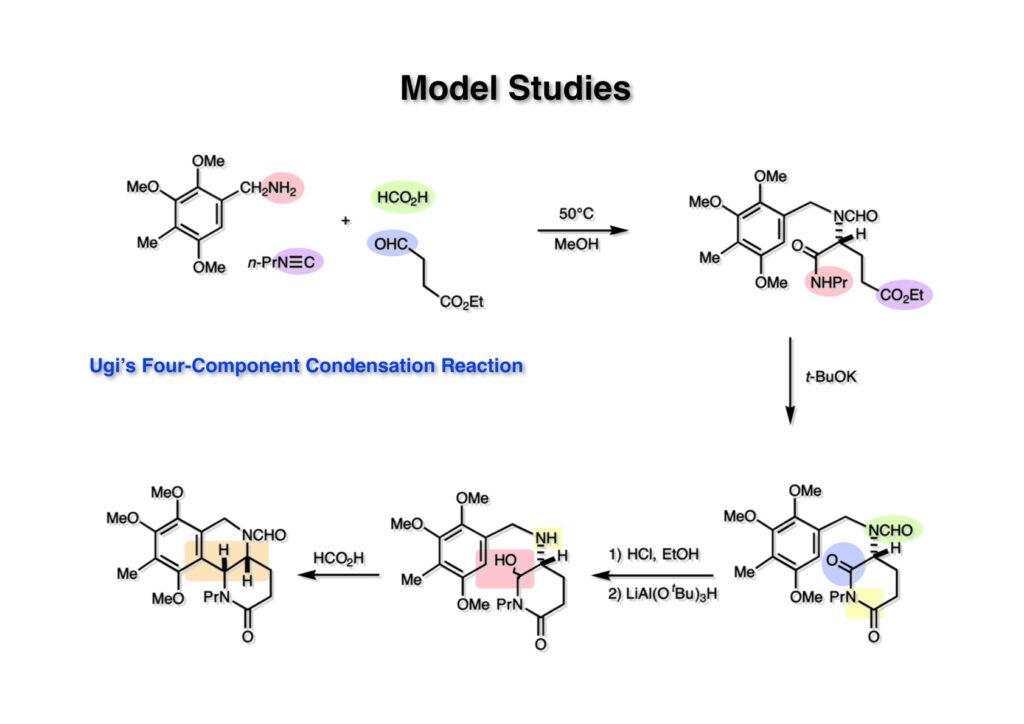
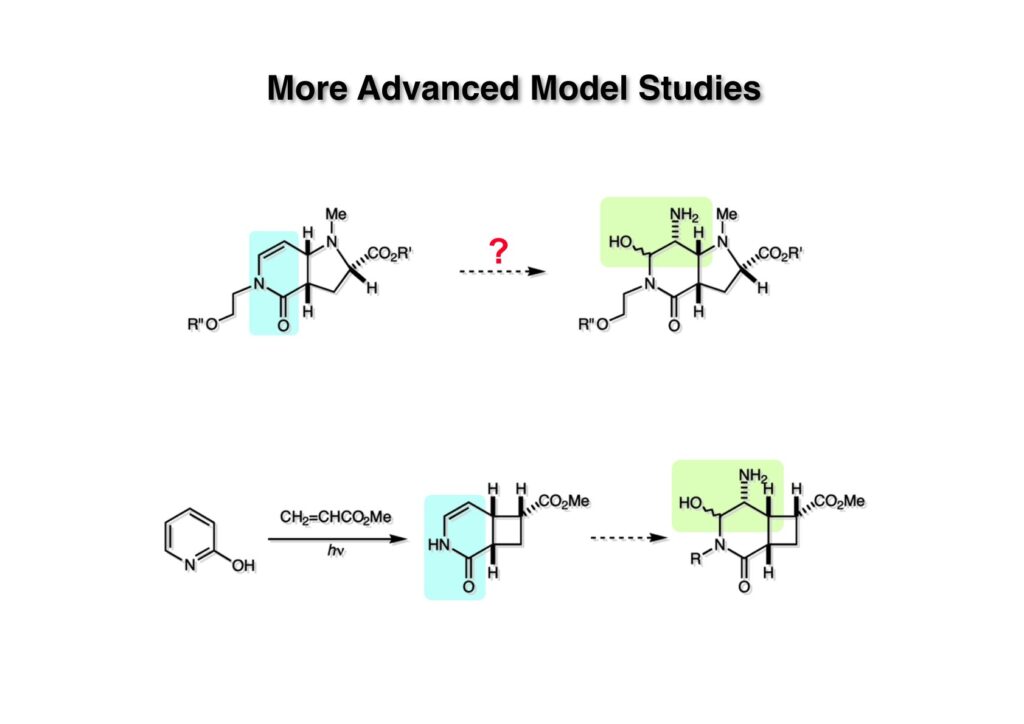
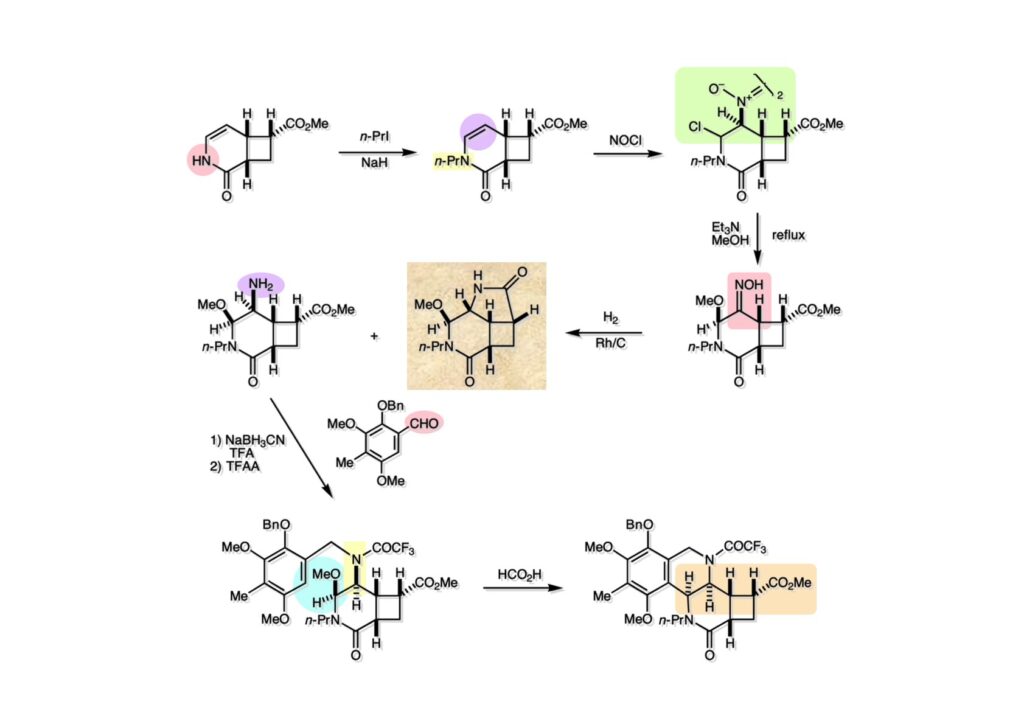
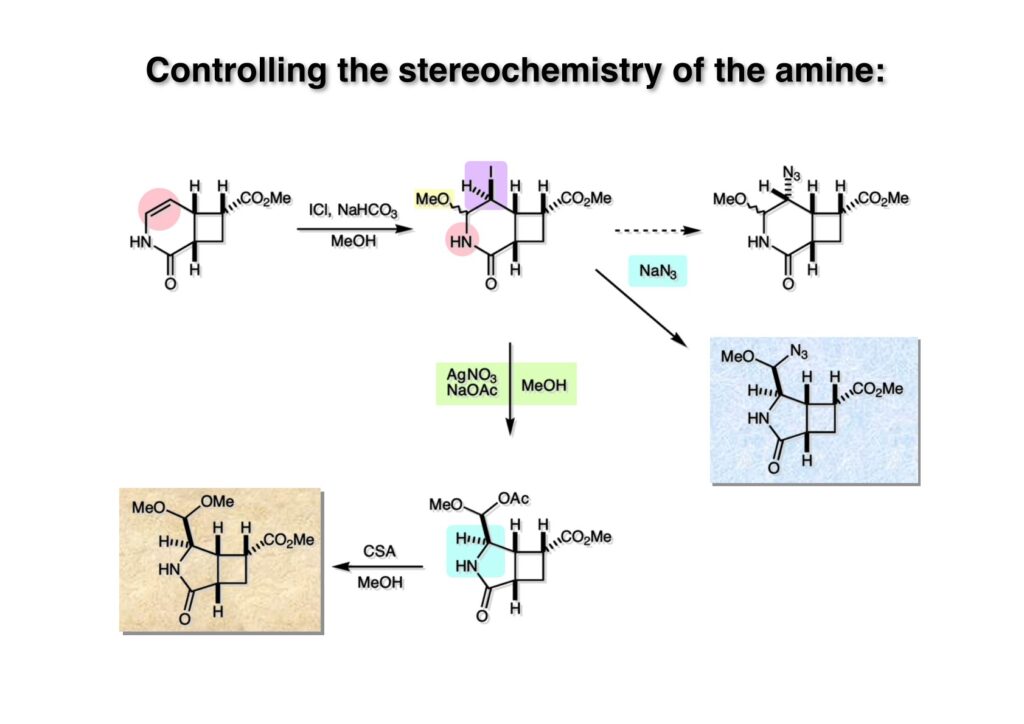
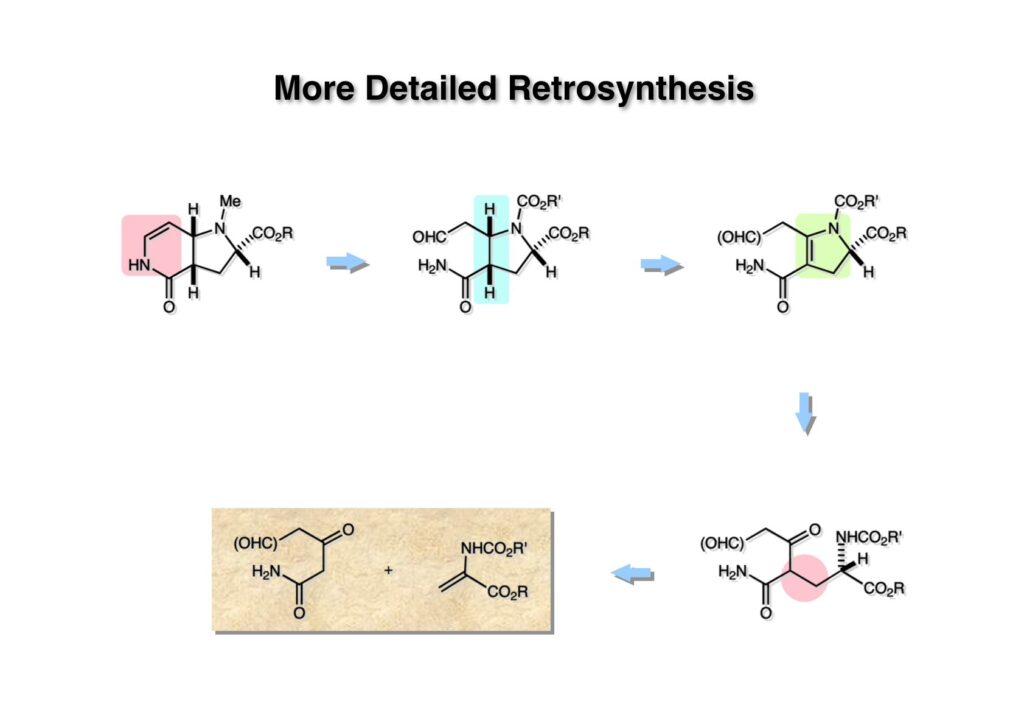
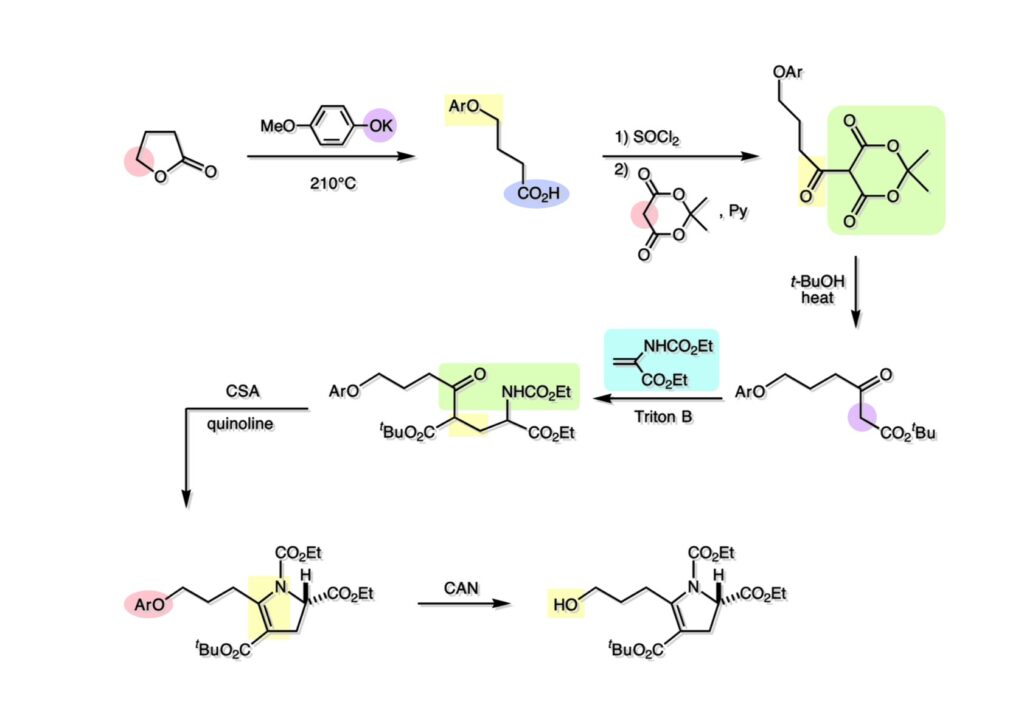
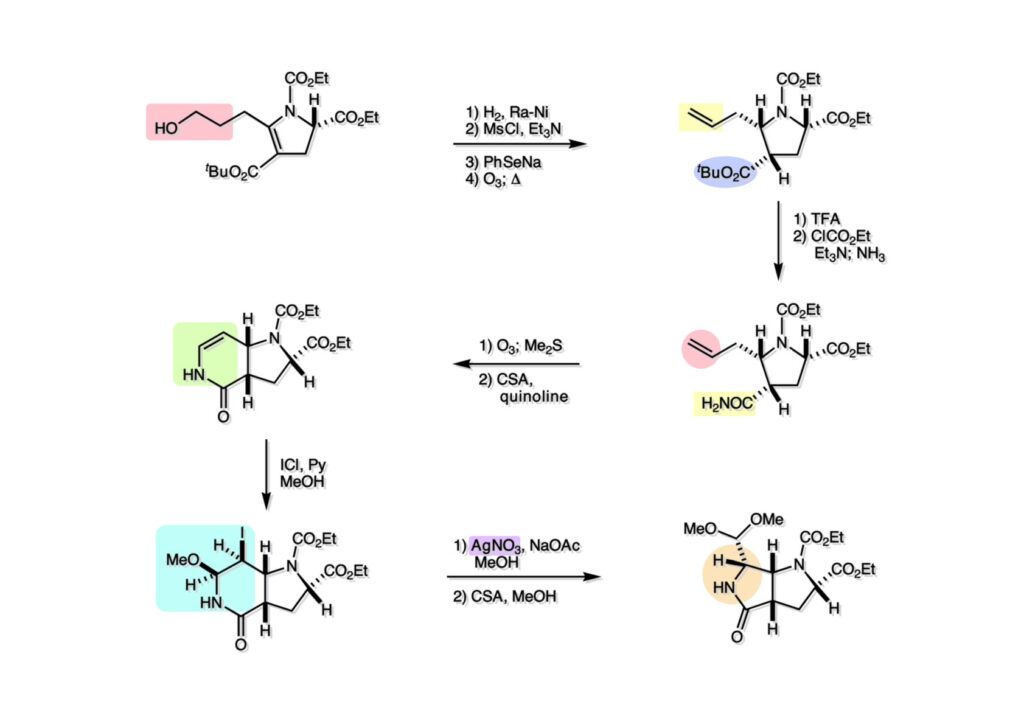
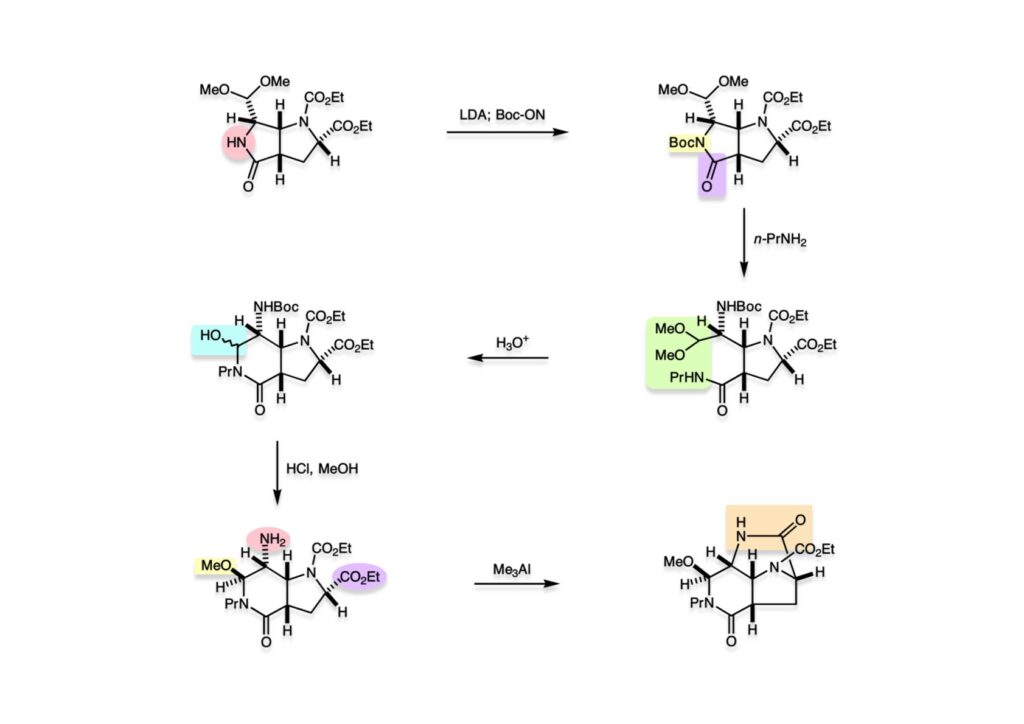
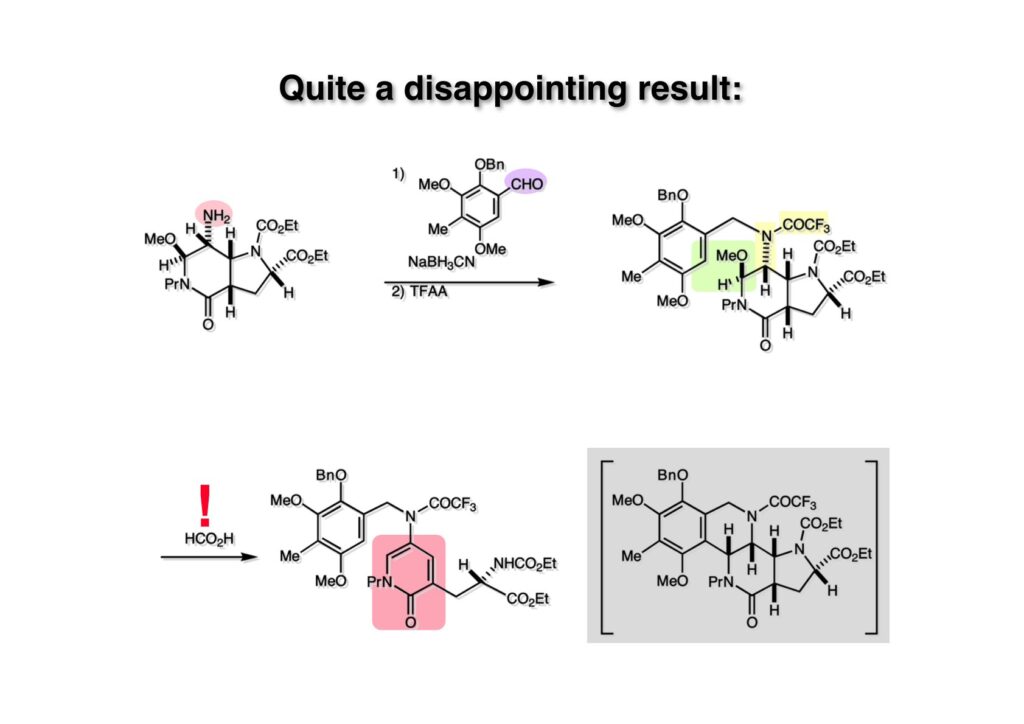
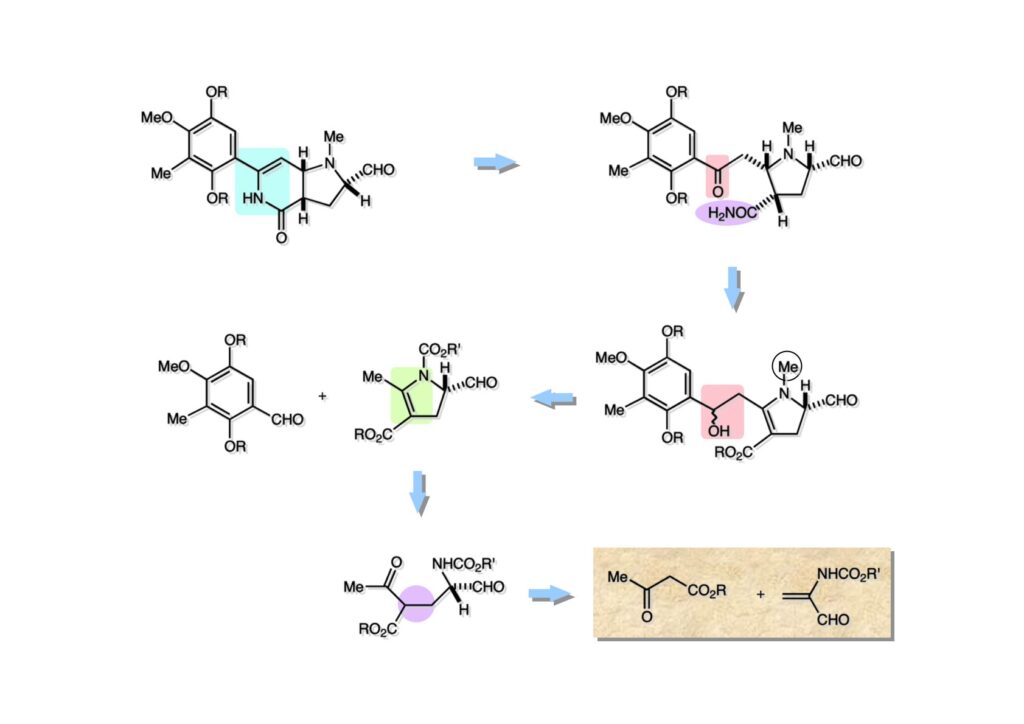
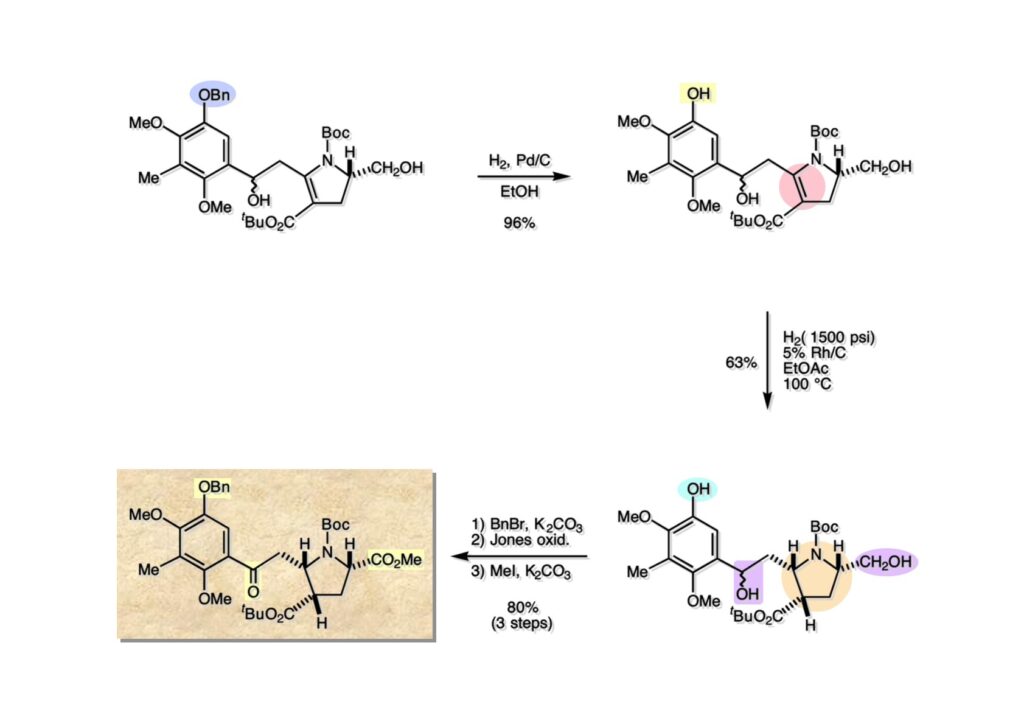
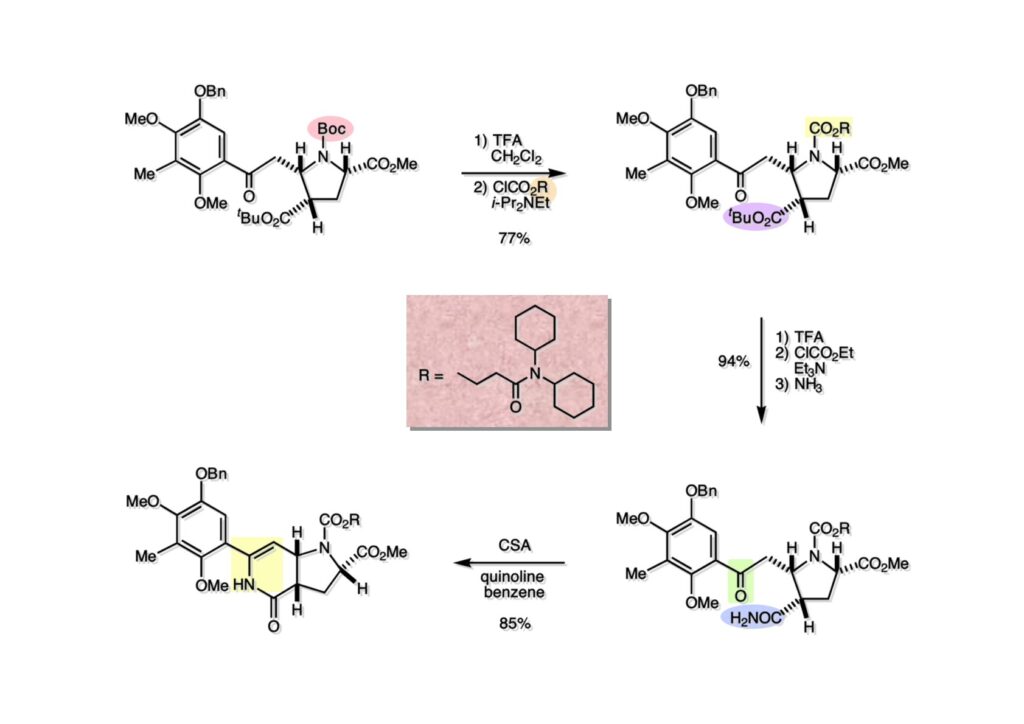
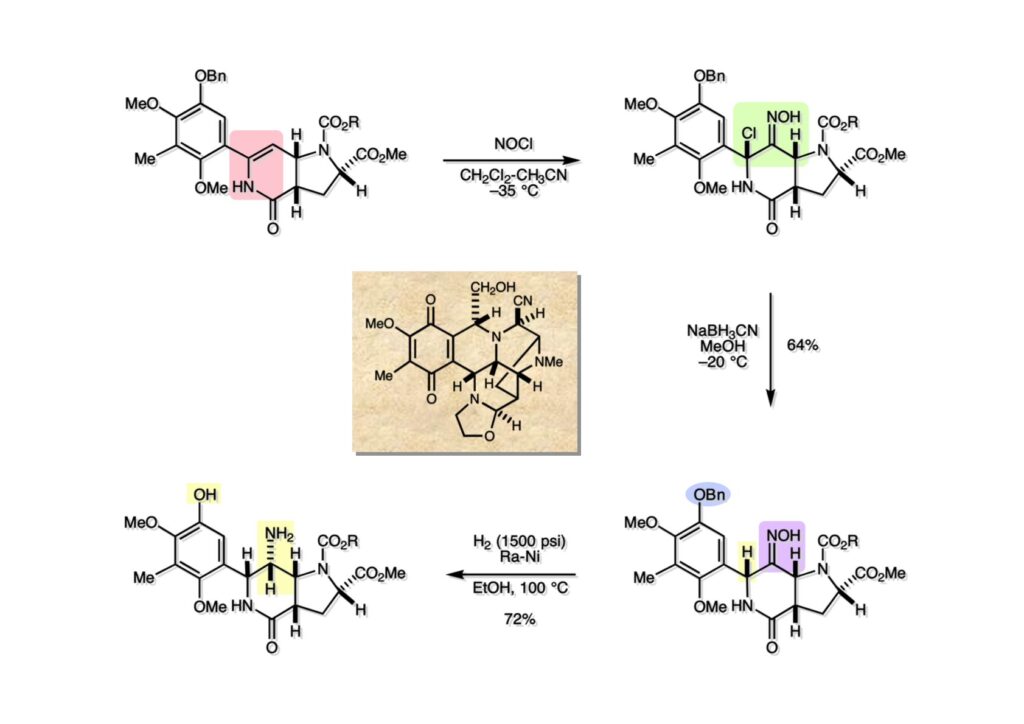
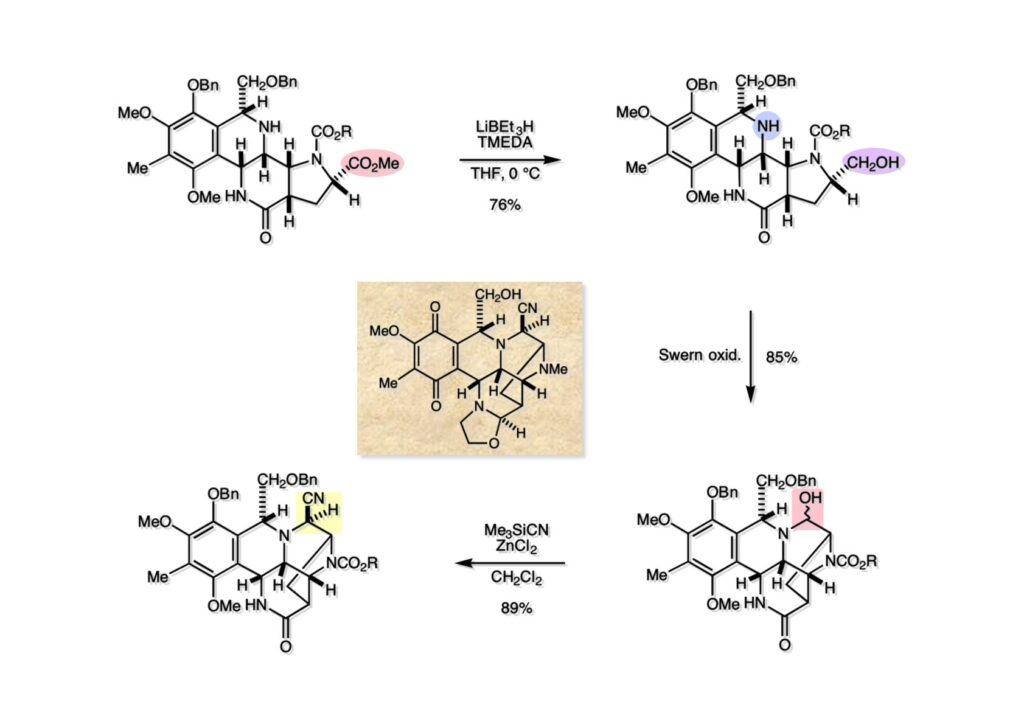
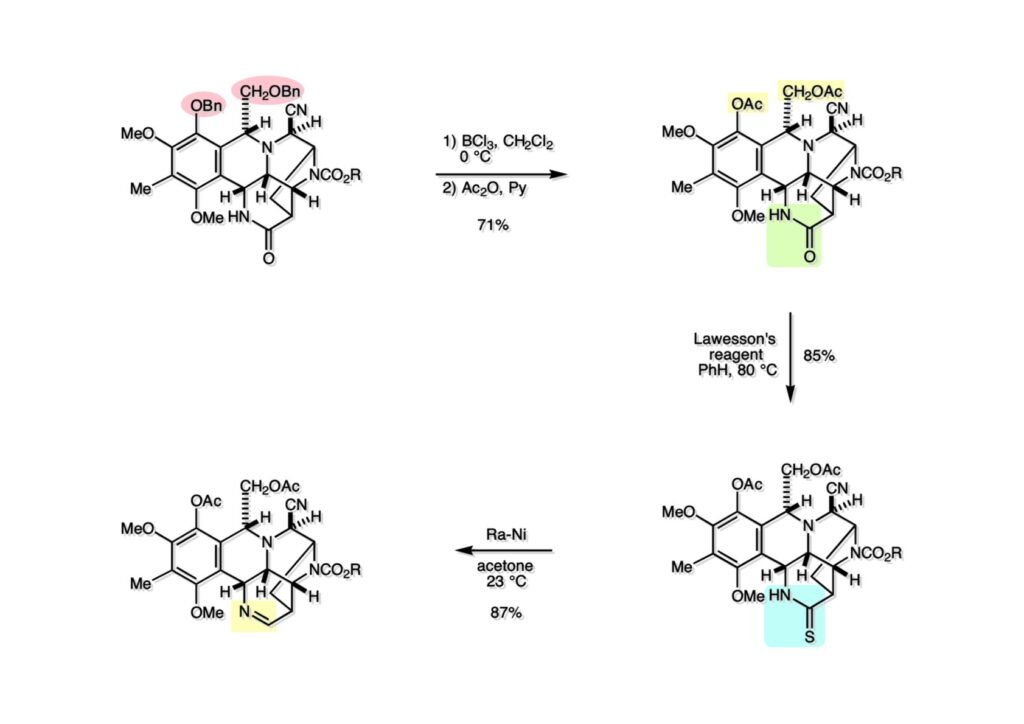
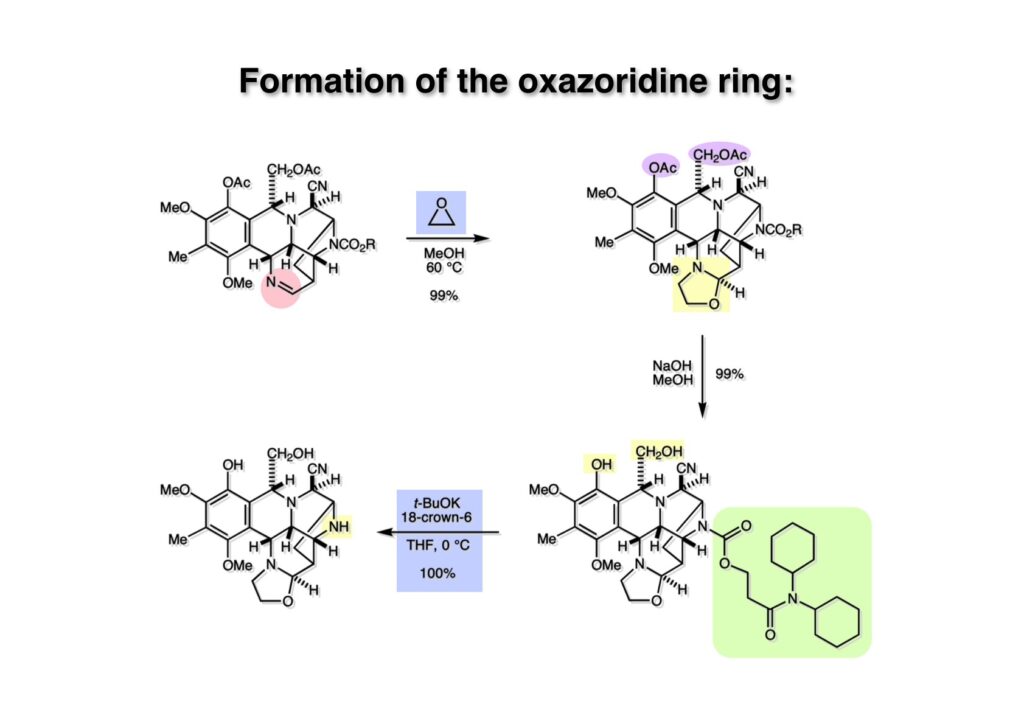
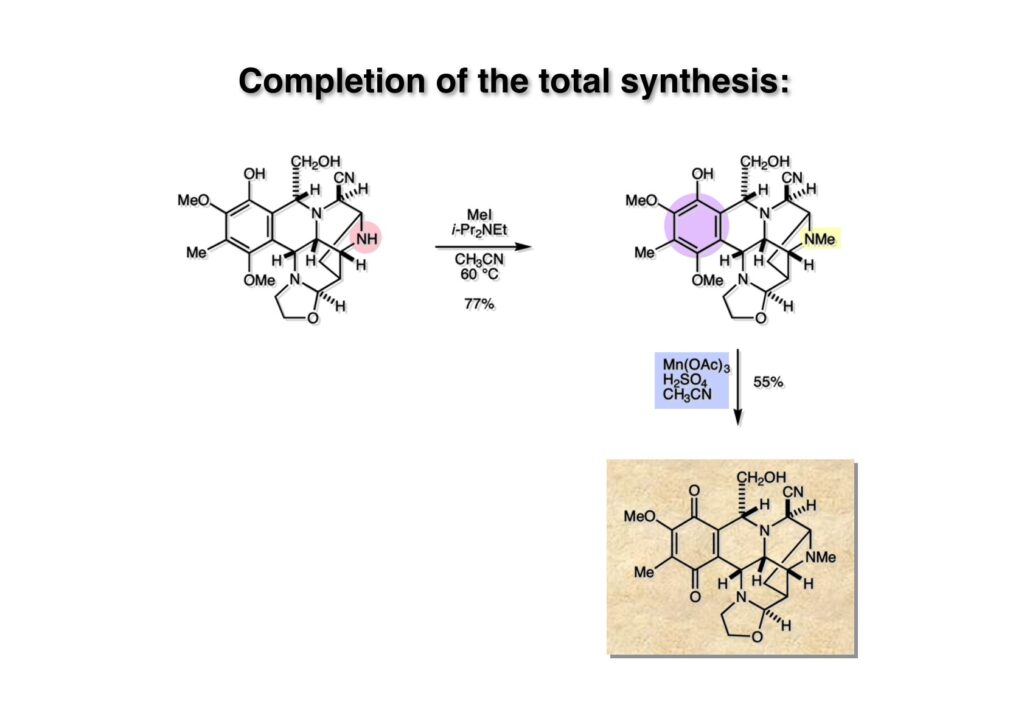
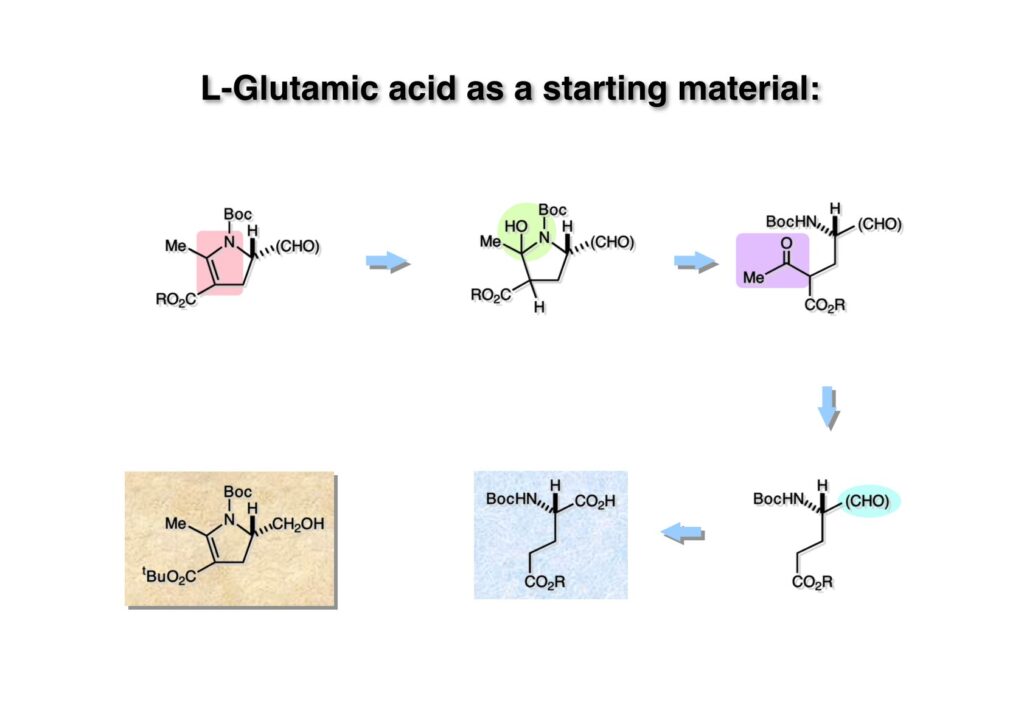
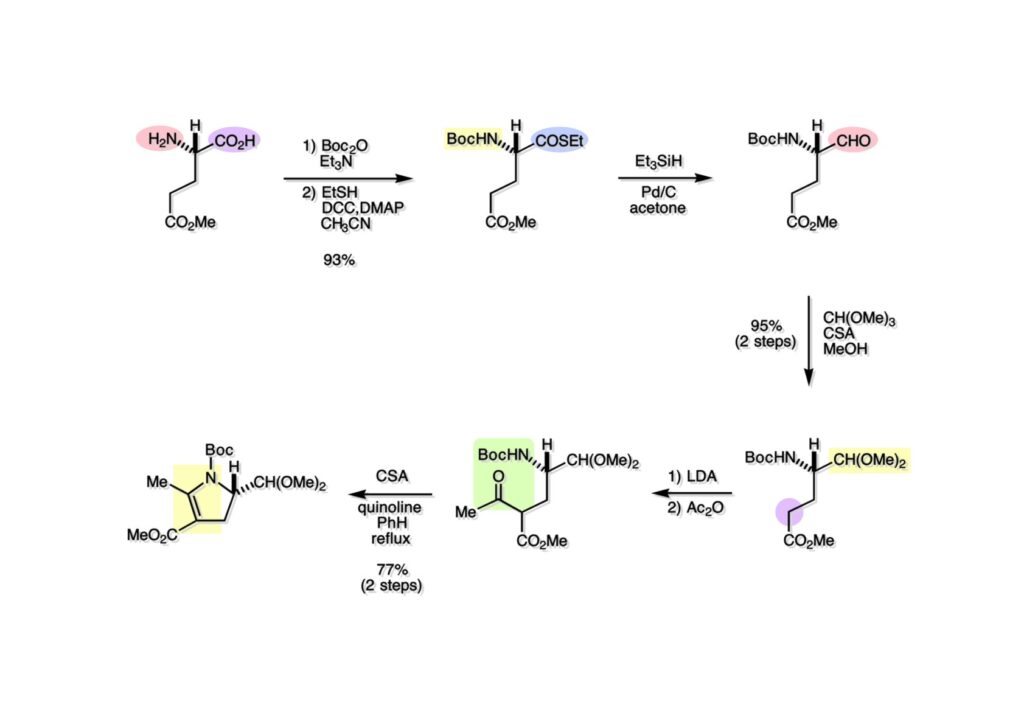
逆合成解析はnaphthyridinomycinを出発点とするがcyanocyclineと同じような化合物である。まずキノンは芳香環として保護し、不安定なヘミアミナールはラクタムにしておけば良い。(1-2) のoxazolidine環はラクタムとアルコールから構築できると考えて (1-3) に導いた。ここで上部のラクタムを開裂させると4環性化合物 (2-2) となる。次に紫色でハイライトした結合はaxyliminium ionを生成して環化すれば良いと考えて (2-1) に変換した。芳香環部分とアミンはケトン (3-1) とアミン (3-2) の還元的アミノ化をすれば良いのでは、と期待した。え、立体化学はどうするの?? そんなのは後で考えれば良いことだ。逆合成はあまり細かいことを気にすると先に進めなくなるよ(と、TFは無責任な忠告をここでしておこう)。アミン部分 (3-2) はエナミド体 (3-3) に酸化的に窒素を入れれば何とかなるだろうと考えて全合成を開始した。 まずモデル実験としてacyliminium ionを生成して3環性化合物ができるかを試してみることにした。例の便利なUgi反応を利用してアミノ酸誘導体 (1-2) を合成し、t -BuOKを用いて6員環イミド (2-3) に導いた。ホルムアミドのままイミドを還元すると位置異性体が生じたので、ホルミル基をHCl-EtOHで外してLiAl(OBu-t )3Hで還元したところ、おそらく試薬のLi+がアミンの孤立電子対に配位したためか、望む還元体 (2-2) のみが得られた。(2-2) をギ酸中で加熱するとスムーズに環化が起こり (2-1) が生成した。おそらく立体化学はcis であると思うが確認はしていない。ついでに言及しておくが、アミンはギ酸中で加熱するとホルムアミドになりやすいので注意が必要である。 単純なモデル実験は成功したが、本番では (1-1) のようなエナミド前駆体からアミン体 (1-2) を合成しなければならない。そこで実際の合成に近いモデル化合物を用いてエナミドからアミンへの変換を行う必要があり、文献調査を始めた。その頃はSciFinderが無い時代だったので図書館でChemical Abstractsのジャングルを彷徨うしか方法がなかった。その結果エナミド体が簡単に得られる文献が見つかった。確か鹿児島大学工学部の先生の論文だったが、2-hydroxypyridin (2-1) とアクリル酸メチルの混合物に光を当てると付加体 (2-2) が30%くらいの収率で得られるという反応だった。実は論文ではCO2Me基はβ配置だったが、反応をやっていくうちにα配置であることが判明した。鹿児島大の先生には間違っているという連絡はしなかったけど。蛇足ながら4員環の1H結合定数は少々トリッキーであることも書き留めておく。 モデル化合物を (1-1) から合成するにあたり、まずラクタムのNHをプロピル化して保護基の代わりにしておいた (1-2)。エナミド体 (1-2) に低温下でNOClのCH2Cl2溶液を滴下していくと、濃い水色が現れてすぐに無色になるという現象が起きた。ニトロソアルカンは青色をしているが、すぐに二量化して無色のazodioxy化合物 (1-3) になる。 これをメタノール中Et3N存在下で加熱するとモノマーに分解し、互変異性を経てオキシム体 (2-3) が得られた。このオキシム (2-3) の還元は容易ではなかったが、Rh/C触媒を用いて高圧下で水添することで2種類の化合物が生じた。主生成物にはメチルエステルが消失し、アミンでもなく、質量分析でラクタム体 (2-2) であることが判明した。ここで光反応の生成物の構造決定が間違っていたことが分かったのだ。本当はこちらのアミンを使って鍵反応である環化を行いたかったのだが、アミンの立体配置が逆の (2-1) を用いて先に進んだ。アルデヒド (3-1) とアミンの還元的アミノ化と続くアミンの保護で (4-1) を得た。これをギ酸処理すると望む4環性の化合物 (4-2) が得られたがtetrahydroisoquinoline環の部分が逆のものであるので安心は出来なかった。 次にやるべき事はα配置のアミンを合成してacyliminium ionを経由した環化が実現できるかを確かめる事だった。今考えると、なぜ (1-1) のエステルを還元してからオキシムの還元をしなかったのか?ちょっと思い出せないが、当然すべき事をやってなかったのが気にはなる。いずれにしても (1-1) の分詞模型を見る限りβ面が開いているので試薬の攻撃はβ面からで、あとは窒素求核剤で立体化学を反転させれば所望のアミンになると思った。I2よりも反応性の高いIClをメタノール中で (1-1) に作用させるとジアステレオマー混合物 (1-2) が得られた。これをNaN3と加熱することで (1-3) が得られると思ったが、実際に生成したのは環縮小された (2-1) であった。ラクタム窒素の隣接基関与の結果である。次に、メタノール中硝酸銀を作用させることで、よりスムーズに (3-2) が得られた。これをメタノール中で酸処理することによりアセタール (3-1) を単一物として得た。 これ以上モデル化合物を扱っても時間の無駄だと思い、前述の立体反転を基軸とした本格的なルート探索に着手した。この時点での目的化合物は (1-1) で、その逆合成解析をここに示す。エナミドを構築するためにはアルデヒドとアミドが必要なので (1-2) が前駆体として欲しい化合物である。側鎖の立体化学をcis に制御するには4置換オレフィン (1-3) の接触還元をすれば良い。ここで注釈を入れるならば、5員環の二重結合の水添は既存の置換基の反対側から起こることがほぼ確実で、トリッキーな6員環とは一線を画する。(1-3) のようなエナミド体を得るにはケトンとウレタンとで環化すれば良いので (2-3) へと導かれる。(2-3) はβ-ケトアミドなので (2-1) と (2-2) のMichael反応で構築できると考えた。ただし、ここで厄介なのは (2-1) のδ位にアルデヒドに変換すべくヘテロ原子を入れておくと、Michael反応の塩基性条件下で脱離する危険性があることだ。 まず4位が保護された4-hydroxybutyric acid (1-3) を合成したが、これは簡単なやり方でできる。Alison Lairdの博士論文によると0.5 molのp -methoxyphenolを丸底フラスコに入れ、次に0.62 molのKOHペレットを入れ、そして1.0 molのγ-ブチロラクトンと攪拌子を入れる。フラスコにエバポレータトラップを取り付けて徐々に210度まで加熱する。この間にKOH中の水はトラップに溜まり、17時間加熱攪拌後に100 mlの水を加えてカリウム塩を溶かしてから希塩酸に注ぎ込むと直ちに結晶の生成物が得られた。濾過、乾燥後65.9 gの (1-3) が得られた(62.7% yield)。勿論、この手抜き実験は私の指示による。(1-3) を酸塩化物に変換後Meldrum酸 (1-4) と反応させると (1-5) が得られる。これをt -BuOH中で加熱すると、アシルケテン中間体を経て β-ケトエステル (2-3) に変換される。この優れた方法は北大薬学部の及川裕二氏と米光宰先生が報告された。及川氏は岸研に留学され帰国された4年後にガンで亡くなられたが、長生きされていたら日本の有機化学の発展に大いに貢献された逸材だった。p -Methoxyphenyl基の除去はCAN酸化で簡単行えることはantibiotic 593Aの全合成で述べたとおりである。 4置換の2-ピロリンの接触還元はRaneyニッケルを用いて高温高圧下で行ったが、後にRh/Al2O3を添加するとより効率的に還元が進行することが分かった。アルコール (1-1) をメシレートに変換し、PhSeNaと反応させてセレナイドにした。これをオゾンでセレノキサイドにして酢酸エチル中で環流してオレフィン (1-2) が得られた。Sulfideやselenideをsulfoxideやselenoxideに変換するには二重結合が無い限りオゾン酸化が便利である。MCPBAなどの過酸と違い酸化され過ぎる心配が無いからであり、後処理も溶媒を留去するだけで良い。次いでt -ブチルエステル (1-2) をTFA処理で外し、混合酸無水物を経てアンモニアを通ずるとアミド (2-2) が得られる。(2-2) の二重結合をオゾン分解し、得られたアルデヒドに弱酸性条件で環化脱水することでエナミド体 (2-1) に導いた。(2-1) を前述の反応に付すことでラクタム体 (3-2) が得られた。 ラクタム (1-1) をN -Boc化し、得られたイミド体 (1-2) をプロピルアミンと加熱すると開環したアミド体 (2-2) が得られた。(2-2) をメタノール中で酸処理しても環化してラクタム (3-1) が得られなかったので、まずアセタールを加水分解してラクタム (2-1) に変換してからHCl/MeOH処理で (3-1) を得た。ここで立体化学を確認するために (3-1) をMe3Alで処理したところ、ラクタム (3-2) が得られたので望む構造を持つことが分かった。 いよいよ重要な環化反応を行うため、アミン (1-1) とアルデヒド (1-2) を還元的アミノ化で連結し、アミンをトリフロロアミド (1-3) として保護した。ところが、(1-3) をギ酸中で加熱しても全く望む環化体 (2-2) は得られず、芳香環化(ピリドン)した (2-1) が得られるのみであった。それだけ立体障害が大きかったということである。結構手間暇かけてここまで来たのにガックリとくる結果であった。まあ、しかし、天然物全合成化学者はこんなことでは負けない。一晩眠れば次のアイデアに着手するだけである。 ここには書かれていない様々なアプローチを試みながら、ほぼ4年の歳月が経った。当時まだ現役であった私がふと見つけた反応がきっかけとなり、これから1年ほどでcyanocycline Aの全合成が完成したが、どのような実験だったかをこのページで説明する。.前述のようにオキシム (1-1) の接触管弦は高温高圧を必要としたので、まずオキシムをNaBH3CNでRNHOHに還元し、その後に別法でN-O結合を還元的に切断してアミンを得ようと考えた。(1-1) と酢酸のメタノール溶液にNaBH3CNを加えたところ、すぐに反応が完結したのでNMRを測定した。何とMeO基が除去されたオキシム (1-3) がきれいに得られてきたのだ。おそらく酸触媒によってメタノールが脱離したニトロソオレフィン (1-2) が生成し、それが共役還元されて (1-3) が生成したと考えられる。これは何を意味するかというと、(2-1) のような化合物をNaBH3CN-acidという条件で還元すると、convex面から還元が起きて、望む立体化学を持つ化合物 (2-2) が得られる可能性が高いということである。 前ページの反応が見つかってから、早速逆合成解析に取り掛かった。ここでもnaphthyridinomycin (1-1) からスタートするが、まず、キノンを芳香環として保護し、ヘミアミナールを開裂して (1-2) に導く。オキサゾリジン環はラクタム (2-2) から構築するとし、次は逆Pictet-Spengler反応で (2-1) にする。(2-1) のアミンはオキシム (3-1) をconvex面から還元すればよく、この化合物はエナミド (3-2) から得られると確信した。(注)ここではアルデヒドのまま逆合成を行っているが、大筋に関係ないからだけで、勿論保護しておかなければならない。 エナミド (1-1) は勿論 (1-2) のケトンとアミドの環化脱水反応で構築できる。(1-2) のピロリジン環はピロリン (2-3) の接触還元で得られるはずである。次は少々飛躍するが、(2-2) のようなα,β-不飽和エステルのアニオンとアルデヒドとの反応は速度論的にはα位で反応するが、平衡反応が成立する場合にはγ位の付加で落ち着くという例がある。従って、(2-3) はアルデヒド (2-1) と不飽和エステル (2-2) という比較的合成容易な化合物に分離できる。(2-2) は開環することで (3-1) へ導かれ、さらにアセト酢酸エステル (3-2) と (3-3) の合成等価体へと簡略化できる。 いよいよモデル実験ではない本格的な全合成の始まりである。(1-3) のようなデヒドロアラニン誘導体は、通常ピルビン酸エステルとカルバメート(H2NCO2R)との酸触媒による加熱脱水縮合で合成されるが、Boc基はその条件に耐えられないのでaza-Wittig反応で合成した。(1-3) と市販のt -butyl acetoacetate (2-1) とのMichael付加反応はどんなに腕の悪い学生でも失敗することがない。つまり生成物 (3-3) が環化していてβ-ケトエステルではないので2度目のMichael付加は起きないからだ。(3-3) の脱水反応は容易に進行してピロリン体 (3-2) を与えた。この段階でエチルエステルをsuper hydrideで選択的に還元してアルコール (3-1) を得た。(3-1) をLDAでエノール化してアルデヒド (4-2) を加えると、付加は進行するもののラクトン化が起きてしまうので、ZnCl2を加えて亜鉛エノレート (4-1) にしてからアルデヒドを加えた。予想通りα位にアルドール型反応が起きたが、徐々にγ位が反応したジアステレオマー混合物 (4-3) に収束していった。亜鉛エノレートを用いるとt- ブチルエステルとのラクトン化は起きなかったが、後の光学活性なcyanocycline Aの全合成では意図的にラクトン環中間体を使った。 次にピロリン (1-1) のオレフィンをRaneyニッケルよりは効率の良いRh/Cを触媒にして水添したが、目的物 (2-2) とベンジル基が還元された (2-2) のcyclohexylmethylエーテルとのほぼ1:1混合物になるという間抜けな結果になった。Rh/Cは芳香環を効率よく還元する触媒であることを頭に入れておかなければならない。仕方ないので (1-1) のベンジル基をPd/Cを使って加水素分解し (1-2) にしてからオレフィンの還元を行って (2-2) を得た。次いでフェノールのベンジル化、2つのアルコールのJones酸化、得られたカルボン酸のメチルエステル化を経て (2-1) に導いた。 さて、化合物 (1-1) のBoc基は前途多難な道を無事に生き残るとは考えられず、酸性、アルカリ性、還元、酸化条件でかなり安定で、架け替える必要のない保護基と交代させる必要があった。そこで、見かけは超ダサいが嵩高いβ-カルボニルオキシプロピオンアミドを保護基とした。まあ、丈夫であることは間違いないが、1H NMRで4 ppmから1 ppmにかけてドヨーンとしたシグナルで占められて見っともないと思ったものだ。(1-1) をTFAのCH2Cl2溶液で処理するとBoc基だけが落ちてくる。通常t -ブチルエステルはTFAそのものを使わないと落ちないので区別は容易である。アミドの窒素にシクロヘキシル基が2つ付いたクロルギ酸エステルを作用させて (1-2) を得た。次にt -ブチルエステルをカルボン酸に変換し、ClCO2Et-Et3Nで混合酸無水物にしてからアンモニア処理してアミド (2-2) を得た。(2-2) をCSA-quinolineでベンゼン中加熱することで環化脱水反応が起きてエナミド体 (2-1) が高収率で得られた。 さて次は本全合成の天王山と言える変換反応である。エナミド (1-1) に-35度でNOClを加えると、今回は立体障害のためかニトロソ体の二量化は起きなかった。次にメタノールを加えてからNaBH3CNで還元するとオキシム (2-2) が主生成物として得られた。オキシム (2-2) の接触還元は高温高圧化で行い、期待通りconvex面から水素化が起きて望むアミン (2-1) を得た。この条件ではベンジルエーテルもフェノールに加水素分解される。 アミノフェノール (1-1) とglycolaldehydeのベンジルエーテル (1-2) のPictet-Spengler反応は中間体のイミニウム塩 (1-3) の分子模型を組んでみるとE -体が優先的にできることは明らかで、実際に高収率でtetrahydroisoquinoline (2-2) が得られた。ここでフェノールをベンジル化して (2-1) に変換した。 メチルエステル (1-1) をsuper hydride (LiBEt3H)でアルコールに還元したが、なぜTMEDAを共存させているかというと、Et3Bが生成物のアミンとcomplexを作るのを防ぐためである。少しではあるが収率が向上した記憶がある。アルコール (1-2) をSwern酸化すると、生じたアルデヒドは直ちにアミンに攻撃されて不安定なヘミアミナール (2-2) が得られる。これを直ちにMe3SiCN-ZnCl2で処理して、ずっと安定なアミノニトリル (2-1) に変換した。 (1-1) の二つのベンジル基を加水素分解で除去しようとすると、フェノール側のベンジル基はあっという間に落ちてくるが、飽和アルコールのベンジル基の加水素分解は通常遅くて、ニトリルの還元も同時進行してしまい収率が低下してします。解決策としては酸性で安定なアミノニトリルなので、BCl3を0度で作用すると脱ベンジル化が完結する。これをアセチル化してジアセテート (1-2) を得た。次のステップはラクタム (1-2) をイミン (2-1) に変換することで、まず、Lawesson試薬を用いてチオラクタム (2-2) に導いた。これにEt3OBF4 (Meerwein試薬)を作用させて得られるチオイミデート (N=C-SEt)をRaney Niで還元しようとしたが、クリーンな結果は得られなかった。そこで (2-2) をアセトンで活性を落としたRaneyニッケルで脱流しようと試みたところ高収率で望むイミン (2-1) が得られた。このイミンは橋頭位の隣りに位置しているためエナミン型になり得ず、安定で扱いやすかった。 イミンをエチレンオキシドと加熱するとoxazolidineが生成するのは既知反応で、(1-1) を大過剰のエチレンオキサイドとメタノール中で加熱すると、ほぼ定量的に望む生成物 (1-2) が得られた。二つのアセテートをまずアルカリ加水分解で除去して (2-2) に変換し、やっとここで「みにくいアヒルの子」におさらばする時がやってきた。(2-2) のアミン保護基は頑丈ではあるが、THF中で18-crown-6存在下、0度でt -BuOKを加えるとアッという間に定量的に除去できた。まあ、そんな事を宣伝しても、こんな特殊な保護基を使ってくれるような物好きなケミストが現れることは期待していない。 アミン (1-1) のメチル化はオキサゾリジンが存在するために還元的アルキル化の方法が使えない。そこでヨウ化メチルを使ったが、メチル化が遅いの何の、大量のHünig baseのメチル化体が生成してしまった。とにかく (1-2) が得られたのでCANを使ってキノンに酸化しようとしたが満足な結果は得られなかった。以前、別のプロジェクトでテトラヒドロイソキノリンの1位を酸化しようとしてMn(OAc)3と加熱したところ、1位は酸化されずにキノンが生成したのを思い出し、これを使ったところ55%の収率でcyanocycline Aが得られた。ここでラセミ体の合成は終了し、次には光学活性なcyanocycline Aを合成することになった。 なぜcyanocycline Aの光学活性体を合成しようと思ったかは、素朴な疑問からだった。確かに抗生物質や環状ペプチドなどでD-アミノ酸が組み込まれている例はたくさんあるが、cyanocycline Aの生合成経路はチロシンとオルニチンからではないかと推察されていた。ということは、中外製薬のX-線解析による絶対構造が正しければD-tyrosineとD-ornithineから生合成されるという、ちょっと不自然な経路となる。これは何かの間違いではないかと確かめる気になったのだ。 ラセミ体の全合成ルートを踏襲するには (1-1) のような光学活性ピロリンを合成すれば良い。 (1-2)を経由して (1-3) に至るのは常識の範囲で、ラセミ体シリーズで用いたβ-ケトエステルのMichael反応は使えないので、(2-3) のアセチル化を試みることにした。当然、全合成の出発物はL-グルタミン酸誘導体 (2-2) ということになる。因みに (2-1) はラセミ体全合成に用いたアルドール型反応の前駆体である。 L-Glutamic acid 5-methyl ester (1-1) のアミノ基をBoc化し、Steglich法でカルボン酸をチオエステル (1-2) に変換した。これを福山還元(この件については後日合成法のページも作って詳述するつもりである)すると高収率でアルデヒド (1-3) が得られた。アミノ酸由来のアルデヒドはラセミ化しやすく、シリカゲルで精製する時にも部分的にラセミ化してしまうという話も聞いた。ここでは精製することなく、溶媒留去で次の反応に使用できるので単離することなくアセタール (2-3) に変換した。次に2等量ちょっとのLDAを用いてエノレートにし、それを過剰な無水酢酸溶液にカニュラで滴下して (2-2) を得た。エノレートに無水酢酸を加えると酸性プロトンが出来るのでエノレートがプロトン化されてしまうので気を付ける必要がある。次いで私たちには常道の方法で環化脱水することでピロリン誘導体 (2-1) が得られる。 ピロリン (1-1) をLDAで脱プロトン化してアルデヒド (1-2) を加えるとラクトン (1-3) が得られる。これをammonolysisと生じた水酸基を酸化することでラセミ体の中間体にほぼ繋ぐことができるので、以後の合成については割愛する。こうやって得られたcyanocycline A (2-1) と中外製薬から頂いた天然物の旋光度ほサインも含めてほぼ一致したので、絶対構造はここに示す通りになることが確定した。Hanessianが論文で示したnaphthyridinomycinの構造も、中外製薬が提示したcyanocycline Aの構造も天然物の鏡像異性体であり、絶対構造が逆だとわかってから中間体も含めていちいち実験ノートに反対の立体化学を書くのに手間取ってしまった(やれやれ)。 中外製薬から何度か実験ができる量のcyanocycline A (1-1) をいただいたので、naphthyridinomycin (1-2) に変換しようと試みた。含水アセトニトリル中で (1-1) を硝酸銀と反応させると、TLC上でわずかに下に見える化合物に変化した。これがnaphthyridinomycinだと思い、飽和食塩水+飽和重曹水+C H2Cl2で分液して目的物と思われる化合物のTLCをチェックしたところ出発物 (1-1) と同じで、1H NMRスペクトルも (1-1) と同じ、つまりノーリアクション!!だった。ここで分かったのは、分液した時にAgCNがNaCNに変換され、分液操作だけでヘミアミナールからアミノニトリルに戻ってしまったということだ。そこで、NaClを使わずに飽和重曹水だけで分液した。Naphthyridinomycinはシリカゲルで精製過程で分解するし、溶媒を留去しても部分的に分解するので、結局精製操作は行わずエバポレータと氷浴を使ってCH2Cl2溶液にエーテルを加えては乾固せぬように溶媒を飛ばし、またエーテルを加えるという操作を何度か繰り返して冷蔵庫に入れておいた。数日後にフラスコを見てみたらルビー色の結晶が出ていたのでX-線解析をしてもらったところnaphthyridinomycin (1-2) であることが判明した。これらの結果は余程の図書館に行かないと読むことができないシリーズ本に収録されているので、いかに私がズボラであるかの証左と言える。