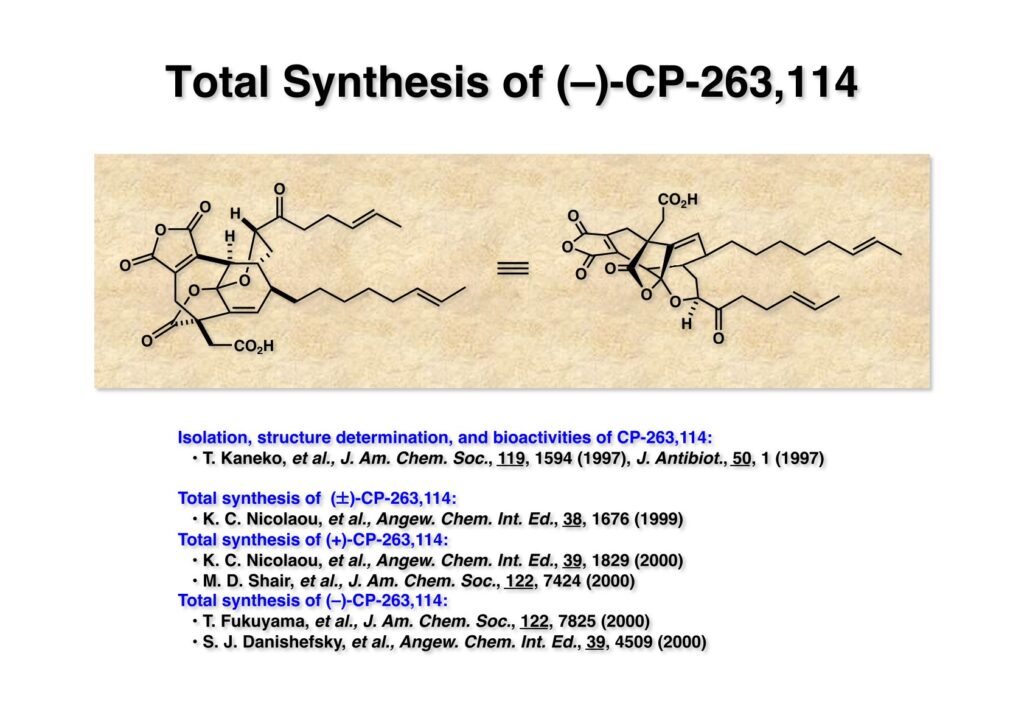
CP-263,114は米国ファイザー社の金子卓史さんが単離構造決定した橋頭位二重結合を有する面白い化合物で、コレステロール低下作用があるらしいが薬にはなっていない。金子さんは岸研で1年間重なっており、飄々とした人柄が懐かしい。彼は広島大学附属高校を卒業してから渡米し、大学は覚えていないが、大学院はミシガン大学のJoe Marino教授のところでPh.D.を取得している。Joeと言えば岸さんとHarvardで重なっていて私も何度かお会いしたことがある。Wikipediaによると、私は「アルカロイド類合成の世界的第一人者」となっているが、裏を返せば窒素が入ってない化合物は合成できない半人前ということになる。それでは私の(わずかに有る)プライドが許さない、ということで金子さんの「窒素の入っていない」化合物を合成してみようではないか、と冗談混じりに語っていたプロジェクトである。1996年に東工大の桑嶋研で修士課程を終えた和泉延明君が研究生としてグループに加わったので早速全合成を開始した。その後、伊東哲志君にも入ってもらい第一世代の全合成を終えた。それから少し後に、いわゆる「福山カップリング」を全合成に適用してみたくなり、伊東君の直接指導の下で林由紀さんが第二世代全合成を完成させた。これもなかなか面白い合成になったと自分では思っている。
“Total Synthesis of (–)-CP-263,114 (Phomoidride B),” N. Waizumi, T. Itoh, and T. Fukuyama, J. Am. Chem. Soc., 122, 7825 (2000).
“A New Synthetic Route to Phomoidride B and Its Derivatives,” Y. Hayashi, T. Itoh, and T. Fukuyama, Org. Lett., 5, 2235 (2003).
 CP-263,114の特徴としては橋頭位の二重結合や反応性の高い無水マレイン酸構造、それにブリッジにケタールラクトンの存在が挙げられる。橋頭位二重結合は9員環の中に含まれているためにBredt則からは十分に安定な構造であると予想される。この特徴ある二重結合は6員環の一部でもあるので当然Diels-Alder反応を用いた構築が考えられる。以前、カリフォルニア大学アーヴァイン校を訪問した時にKen Shea教授とディスカッションしたことがある。彼は有機合成とポリマー合成の両方に興味を持っていたが、分子内Diels-Alder反応を使って橋頭位二重結合を種々合成していたので、逆合成を考えた時にすぐに彼のケミストリーが頭に浮かんだ。
CP-263,114の特徴としては橋頭位の二重結合や反応性の高い無水マレイン酸構造、それにブリッジにケタールラクトンの存在が挙げられる。橋頭位二重結合は9員環の中に含まれているためにBredt則からは十分に安定な構造であると予想される。この特徴ある二重結合は6員環の一部でもあるので当然Diels-Alder反応を用いた構築が考えられる。以前、カリフォルニア大学アーヴァイン校を訪問した時にKen Shea教授とディスカッションしたことがある。彼は有機合成とポリマー合成の両方に興味を持っていたが、分子内Diels-Alder反応を使って橋頭位二重結合を種々合成していたので、逆合成を考えた時にすぐに彼のケミストリーが頭に浮かんだ。
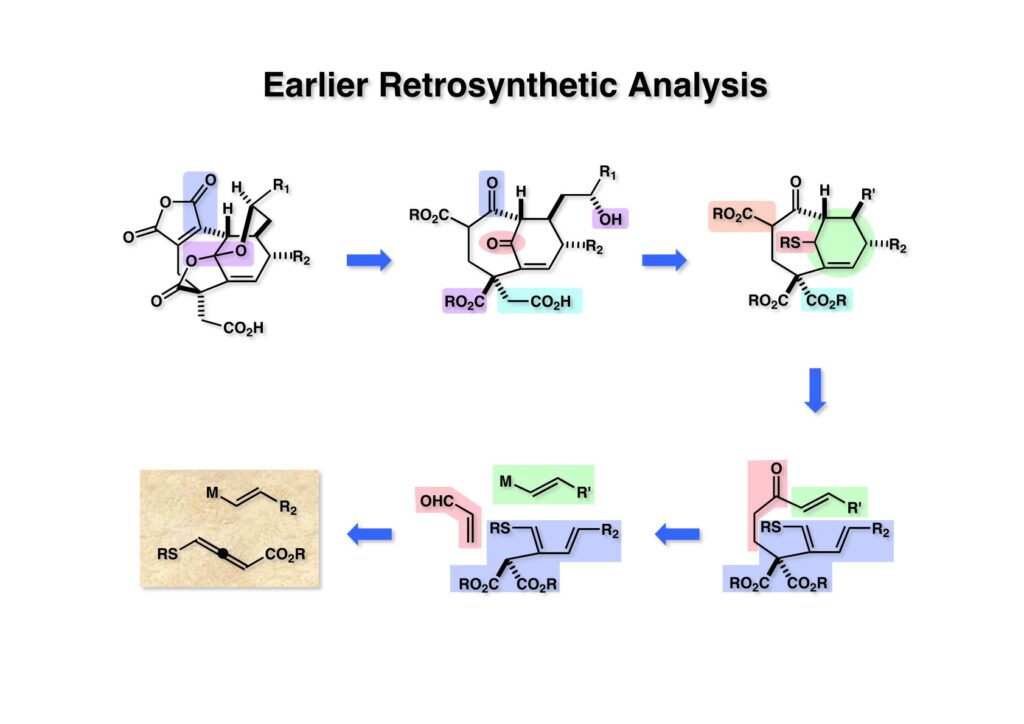 取り敢えずザックリ考えた逆合成解析をこのページに示す。(1-1) のマレイン酸無水物はβ-ケトエステルをエノールトリフレートに変換してPd触媒を使ったカルボニル化を実行すれば良いと考えた。ケタールラクトンは加アルコール分解すればエステル-ケトン-アルコールとなる。(1-2) の四級炭素は不斉合成に手間がかかりそうなのでマロン酸エステルにしておいた方が便利である。ケトンはPummerer転位で構築するならば (1-3) のようなサルファイドに簡略化できる。(1-3) のretro-Diels-Alder反応で導かれるのは (2-3) となる。(2-3) の構築には3つに色分けした部分に分けることができる。マロン酸エステルからはアクロレインへのMichael付加が考えられるし、得られた化合物のアルデヒドにカルバニオンを付加-酸化すれば (2-3) になる。(2-2) のジエン部分の構築はアレンエステル (2-1) への共役付加を行えば立体化学の制御が容易になると考えられた。
取り敢えずザックリ考えた逆合成解析をこのページに示す。(1-1) のマレイン酸無水物はβ-ケトエステルをエノールトリフレートに変換してPd触媒を使ったカルボニル化を実行すれば良いと考えた。ケタールラクトンは加アルコール分解すればエステル-ケトン-アルコールとなる。(1-2) の四級炭素は不斉合成に手間がかかりそうなのでマロン酸エステルにしておいた方が便利である。ケトンはPummerer転位で構築するならば (1-3) のようなサルファイドに簡略化できる。(1-3) のretro-Diels-Alder反応で導かれるのは (2-3) となる。(2-3) の構築には3つに色分けした部分に分けることができる。マロン酸エステルからはアクロレインへのMichael付加が考えられるし、得られた化合物のアルデヒドにカルバニオンを付加-酸化すれば (2-3) になる。(2-2) のジエン部分の構築はアレンエステル (2-1) への共役付加を行えば立体化学の制御が容易になると考えられた。
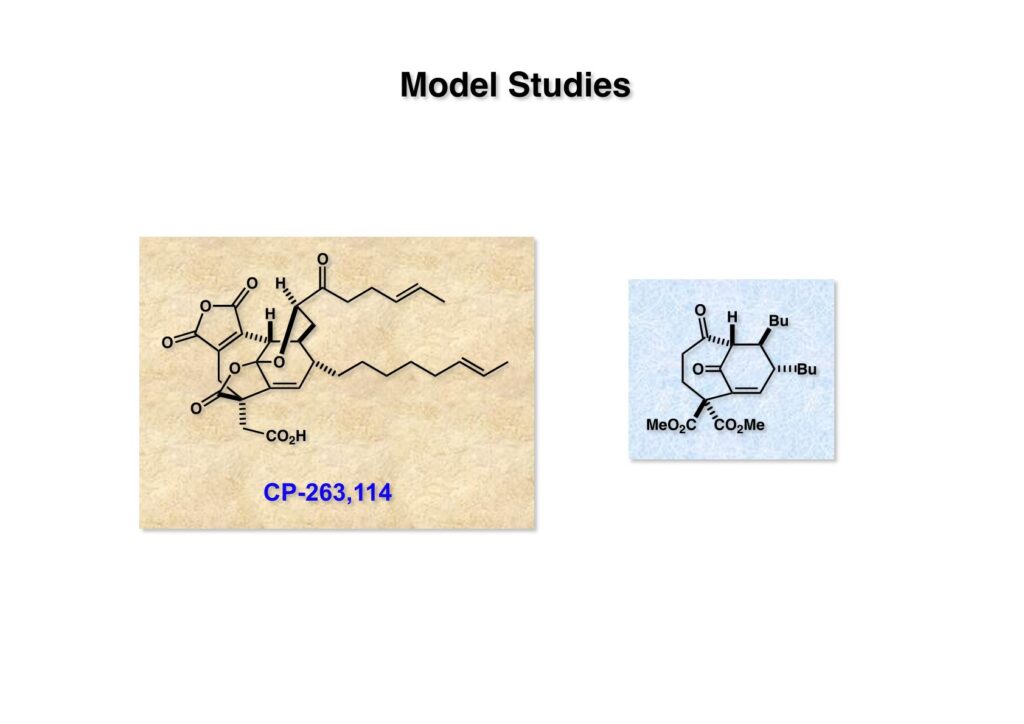 取り敢えず逆合成解析に基づいて側鎖をブチル基にしたモデル化合物 (1-2) を合成できるかどうかを確かめてみることにした。
取り敢えず逆合成解析に基づいて側鎖をブチル基にしたモデル化合物 (1-2) を合成できるかどうかを確かめてみることにした。
 アレンエステル (2-2) の合成は比較的簡単で、市販のプロパルギルブロマイド (1-1) をサルファイド (1-2) に変換し、アセチレン末端をBuLiでアニオンにしてClCO2Meを加えるとエステル (2-1) が得られる。これをDBUで処理するとアセチレンが異性化してアレン体 (2-2) が生成するが、少し不安定なため精製せずに次の反応に供した。
アレンエステル (2-2) の合成は比較的簡単で、市販のプロパルギルブロマイド (1-1) をサルファイド (1-2) に変換し、アセチレン末端をBuLiでアニオンにしてClCO2Meを加えるとエステル (2-1) が得られる。これをDBUで処理するとアセチレンが異性化してアレン体 (2-2) が生成するが、少し不安定なため精製せずに次の反応に供した。
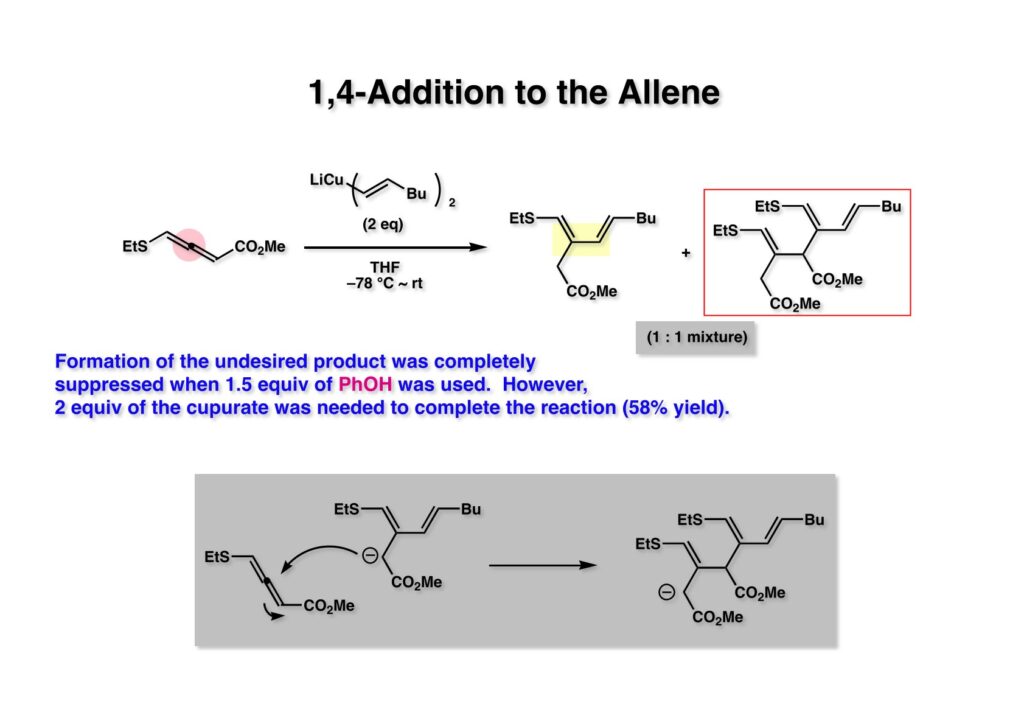 アレンエステル (1-1) にcuprate (1-2) を加えたところ目的物 (1-3) とさらにアレンエステルに共役付加した化合物 (1-4) との1:1混合物が得られてきた。そこでleinamycinの全合成に使ったフェノールを共存させる条件を用いたところ (1-4) の生成は抑制することが出来たが2当量のcuprate (1-2) を必要とした。
アレンエステル (1-1) にcuprate (1-2) を加えたところ目的物 (1-3) とさらにアレンエステルに共役付加した化合物 (1-4) との1:1混合物が得られてきた。そこでleinamycinの全合成に使ったフェノールを共存させる条件を用いたところ (1-4) の生成は抑制することが出来たが2当量のcuprate (1-2) を必要とした。
 そこで和泉君は桑嶋研で開発されたalkenylcopper-Me3SiCl-HMPAという条件を使うことにした。報告された条件では好結果が得られなかったが過剰のMe2Sを加えてalkenylcopper (1-2) の溶解度を増したところ良好な収率で目的物 (1-3) が得られた。ジエンの立体化学はアレンエステル (2-1) のような化合物ではEtS基の立体障害を避けながら反対側から付加が起きるので (1-2) 当然目的物 (1-3) が得られる。(1-3) のようなジエンに分子間Diels-Alder反応を行う場合、Z体になると共平面性 (coplanarity) が保てなくなり反応が進行しにくくなる。分子内Diels-Alder反応ではその限りではないが、E体であるに越したことはない。
そこで和泉君は桑嶋研で開発されたalkenylcopper-Me3SiCl-HMPAという条件を使うことにした。報告された条件では好結果が得られなかったが過剰のMe2Sを加えてalkenylcopper (1-2) の溶解度を増したところ良好な収率で目的物 (1-3) が得られた。ジエンの立体化学はアレンエステル (2-1) のような化合物ではEtS基の立体障害を避けながら反対側から付加が起きるので (1-2) 当然目的物 (1-3) が得られる。(1-3) のようなジエンに分子間Diels-Alder反応を行う場合、Z体になると共平面性 (coplanarity) が保てなくなり反応が進行しにくくなる。分子内Diels-Alder反応ではその限りではないが、E体であるに越したことはない。
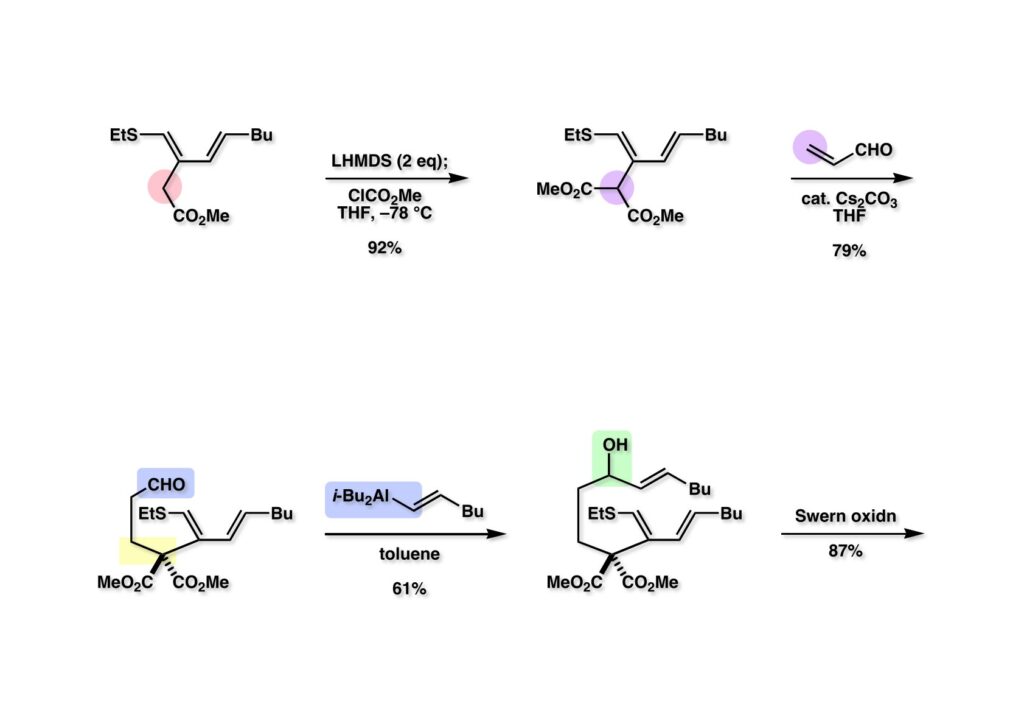 (1-1) のエステルをマロネートに変換するため2当量のLHMDSを用いて脱プロトン化し、ClCO2Meを加えて (1-2) を得た。以前にも言及したが、生成物が出発物よりも脱プロトン化されやすい場合は念の為にも過剰量の塩基を用いるか、カルバニオンを過剰の反応性の高い試薬に加える必要がある (inverse addition) 。得られたマロネート (1-2) はCs2CO3存在下アクロレイン (1-3) と容易に反応して (2-1) を与えた。次に1-hexyneにDIBALを作用 (hydroalumination) させたhexenylaluminum (2-2) をアルデヒド (2-1) に付加させて (2-3) を得た。これをSwern酸化してケトン (次ページ 1-1) に変換した。
(1-1) のエステルをマロネートに変換するため2当量のLHMDSを用いて脱プロトン化し、ClCO2Meを加えて (1-2) を得た。以前にも言及したが、生成物が出発物よりも脱プロトン化されやすい場合は念の為にも過剰量の塩基を用いるか、カルバニオンを過剰の反応性の高い試薬に加える必要がある (inverse addition) 。得られたマロネート (1-2) はCs2CO3存在下アクロレイン (1-3) と容易に反応して (2-1) を与えた。次に1-hexyneにDIBALを作用 (hydroalumination) させたhexenylaluminum (2-2) をアルデヒド (2-1) に付加させて (2-3) を得た。これをSwern酸化してケトン (次ページ 1-1) に変換した。
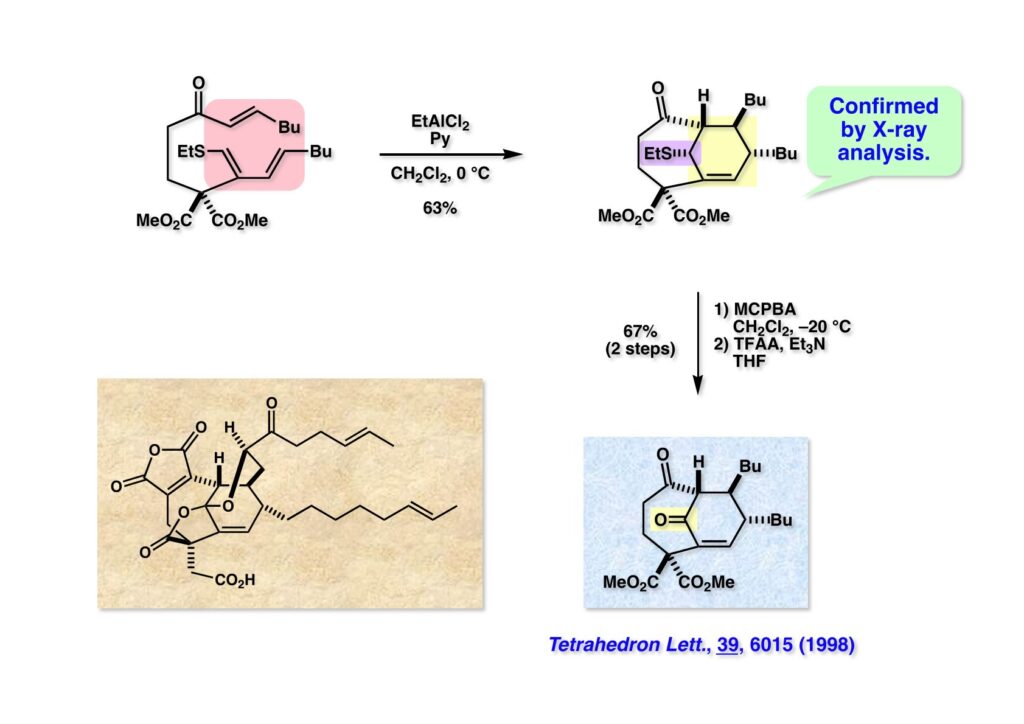 (1-1) の分子内Diels-Alder反応はルイス酸 (EtAlCl2) を加えることで推進され目的物 (1-2) がまずまずの収率で得られた。ここで少量のピリジンを添加しないとビニルサルファイドが異性化した副生物が得られてしまう。(1-2) の構造はX線解析で決定した。続くPummerer転位も問題なく進行してケトン (2-2) が得られた。無水酢酸の代わりに無水トリフロロ酢酸を用いると0度でも反応が進行し、反応終了後に重曹水を加えるとトリフロロアセテートが加水分解されてケトンが容易に得られる。これまでのモデル実験でCP-263,114 (2-1) 全合成への足場がかなり固まったが、光学活性体の全合成にはまだハードルが高かった。
(1-1) の分子内Diels-Alder反応はルイス酸 (EtAlCl2) を加えることで推進され目的物 (1-2) がまずまずの収率で得られた。ここで少量のピリジンを添加しないとビニルサルファイドが異性化した副生物が得られてしまう。(1-2) の構造はX線解析で決定した。続くPummerer転位も問題なく進行してケトン (2-2) が得られた。無水酢酸の代わりに無水トリフロロ酢酸を用いると0度でも反応が進行し、反応終了後に重曹水を加えるとトリフロロアセテートが加水分解されてケトンが容易に得られる。これまでのモデル実験でCP-263,114 (2-1) 全合成への足場がかなり固まったが、光学活性体の全合成にはまだハードルが高かった。
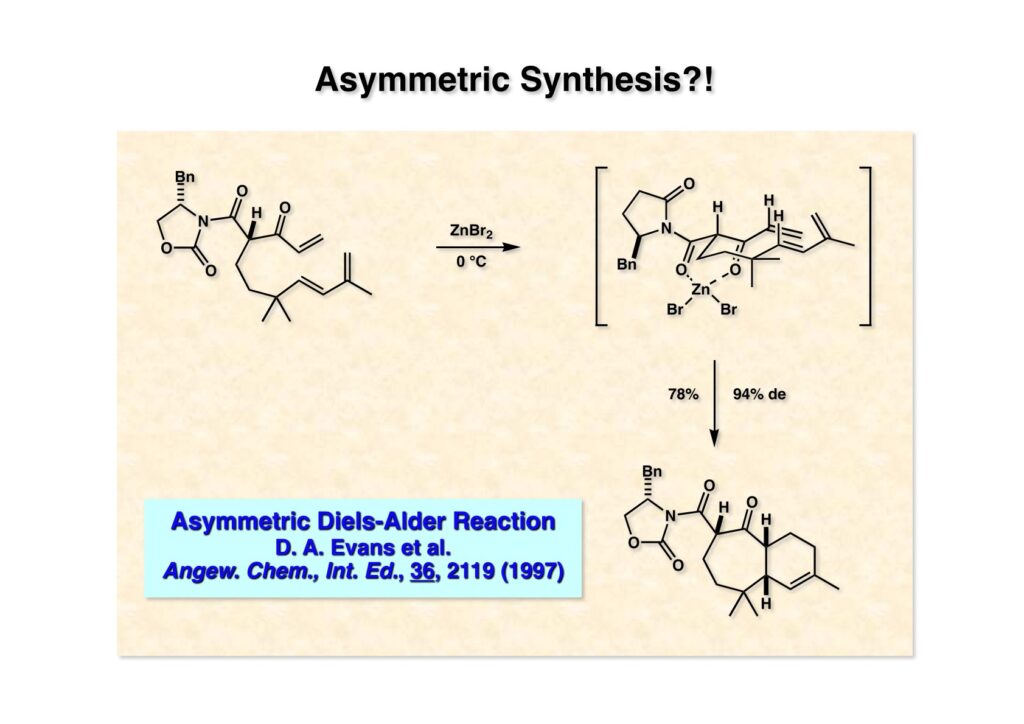 丁度その頃ハーバード大学で講演する機会があり、Dave Evans教授と話をしていたところ分子内不斉Diels-Alder反応が起きるということを知った。(1-1) のようなEvansのキラル補助基がβ-ケトカルボニル体の両カルボニル基の安定配座が直交しているために容易にエピメリ化しないそうだ。(1-1) は実際ZnBr2存在下に反応して94% deで (2-1) を与えている。これは我々のCP全合成にも適用できると思い、帰国後に合成計画を若干変更した。
丁度その頃ハーバード大学で講演する機会があり、Dave Evans教授と話をしていたところ分子内不斉Diels-Alder反応が起きるということを知った。(1-1) のようなEvansのキラル補助基がβ-ケトカルボニル体の両カルボニル基の安定配座が直交しているために容易にエピメリ化しないそうだ。(1-1) は実際ZnBr2存在下に反応して94% deで (2-1) を与えている。これは我々のCP全合成にも適用できると思い、帰国後に合成計画を若干変更した。
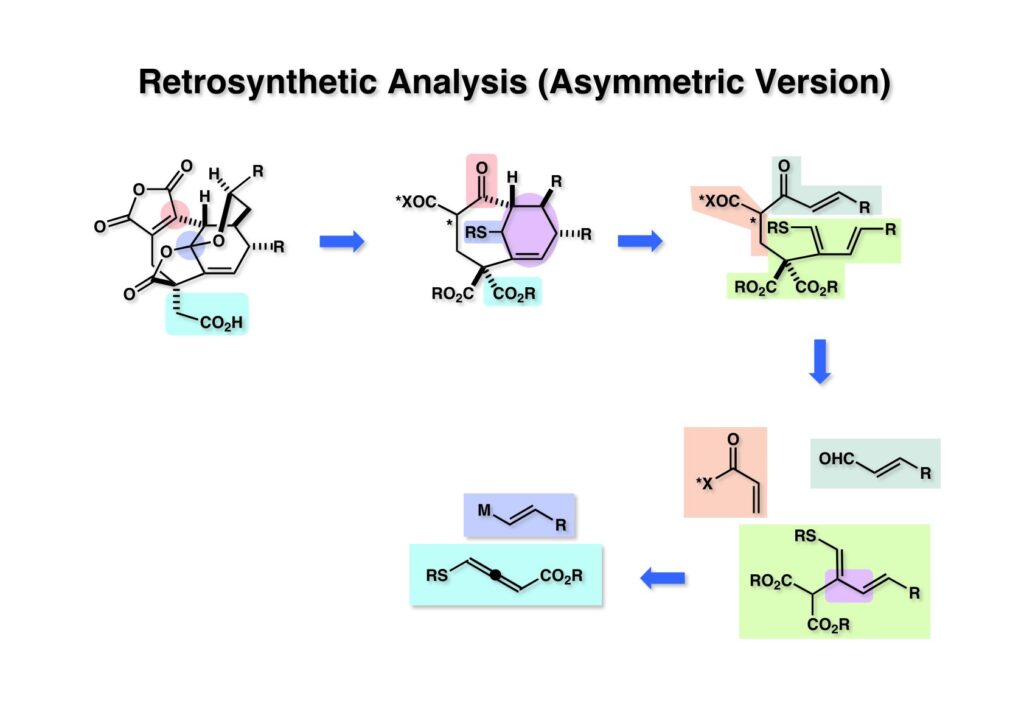 この逆合成解析でお分かりのように、(2-2) のマロネートのMichael反応相手をEvansのキラル補助基を用いたアクリル酸誘導体をしたくらいで、それほど大きな変更を加えたものではない。
この逆合成解析でお分かりのように、(2-2) のマロネートのMichael反応相手をEvansのキラル補助基を用いたアクリル酸誘導体をしたくらいで、それほど大きな変更を加えたものではない。
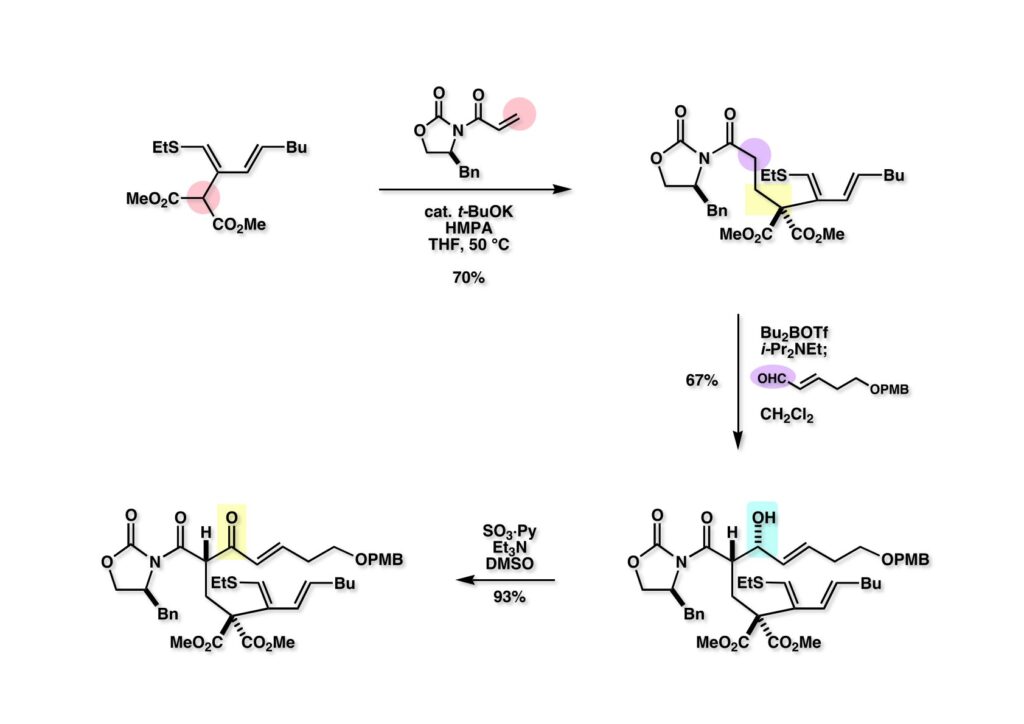 CP-263,114の絶対配置は決定されていなかったので、より安いL-phenylalanineから合成したEvansキラル補助基にアクリル酸を縮合した (1-2) を用いた。NicolaouもShairもent-CP-263,114を合成してしまったが、我々はコイントスに勝って天然物そのものの合成に成功した。
CP-263,114の絶対配置は決定されていなかったので、より安いL-phenylalanineから合成したEvansキラル補助基にアクリル酸を縮合した (1-2) を用いた。NicolaouもShairもent-CP-263,114を合成してしまったが、我々はコイントスに勝って天然物そのものの合成に成功した。
マロネート (1-1) の (1-2) へのMichael付加は期待通りに進行して (1-3) を与えた。Evansのアルドール反応はそれまで実行したことは無かったが、Bu 2BOTfさえ無事に作ってしまえば問題なく進行することが分かった。新品のBu3BとTfOHを使えば大丈夫ということだった。(2-2) を文献通りParikh-Doering酸化を行ってケトン (2-1) が得られた。
 このページのタイトルではAsymmetric Diels-Alder Rxnと書いてあるが、正確にはDiastereoselective Diels-Alder Rxnとすべきであり、ついつい光学活性な目的物を合成するときにasymmetricという言葉を使ってしまうので注意すべきである。(1-1) をルイス酸 (ZnCl2·Et2O) 存在下で室温放置すると分子内Diels-Alder反応が進行して目的とする化合物 (2-2) のみが得られた。ビシクロ体 (2-2) の構造についてはNOE実験 (2-1) でCP-263,114と同じであることを確認した。
このページのタイトルではAsymmetric Diels-Alder Rxnと書いてあるが、正確にはDiastereoselective Diels-Alder Rxnとすべきであり、ついつい光学活性な目的物を合成するときにasymmetricという言葉を使ってしまうので注意すべきである。(1-1) をルイス酸 (ZnCl2·Et2O) 存在下で室温放置すると分子内Diels-Alder反応が進行して目的とする化合物 (2-2) のみが得られた。ビシクロ体 (2-2) の構造についてはNOE実験 (2-1) でCP-263,114と同じであることを確認した。
 次に (1-1) をモデル化合物として無水マレイン酸構造を構築することにした。Evansの不斉補助基を除去する方法は化合物によってtrickyで無造作にやるとオキサゾリジノンのカルボニル基が攻撃されてしまう。ここでもメチルエステルに変換するのにEtSLiを使ってチオエステルにし、それをK2CO3/MeOHで加メタノール分解することでメチルエステル (1-2) を得た。このβ-ケトエステルも2つのカルボニル基が平面に近い配座を取らないためにα-Hの脱プロトン化が簡単でなくLHMDSを使わなければならなかった。エノレートにTF2Oを加えてエノールトリフレートに変換してからMCPBAでサルファイドを酸化して (2-2) を得て、続くPummerer転位でケトン (2-1) に変換された。
次に (1-1) をモデル化合物として無水マレイン酸構造を構築することにした。Evansの不斉補助基を除去する方法は化合物によってtrickyで無造作にやるとオキサゾリジノンのカルボニル基が攻撃されてしまう。ここでもメチルエステルに変換するのにEtSLiを使ってチオエステルにし、それをK2CO3/MeOHで加メタノール分解することでメチルエステル (1-2) を得た。このβ-ケトエステルも2つのカルボニル基が平面に近い配座を取らないためにα-Hの脱プロトン化が簡単でなくLHMDSを使わなければならなかった。エノレートにTF2Oを加えてエノールトリフレートに変換してからMCPBAでサルファイドを酸化して (2-2) を得て、続くPummerer転位でケトン (2-1) に変換された。
 エノールトリフレート (1-1) のPd触媒によるカルボニル化は少々特殊なホスフィンリガンドを用いて比較的低収率ながらも目的とするマレイン酸エステル (1-2) を得ることができた。ここではメチルエステルを加水分解してマレイン酸無水物に変換するところまでは行わず、ケタールラクトンの構築をやってみた。PMB (p-methoxybenzyl) 基の脱保護はMe3SiSiMe3とI2でMe3SiIを生成させて行ったところ目的物 (2-2) が直接得られた。
エノールトリフレート (1-1) のPd触媒によるカルボニル化は少々特殊なホスフィンリガンドを用いて比較的低収率ながらも目的とするマレイン酸エステル (1-2) を得ることができた。ここではメチルエステルを加水分解してマレイン酸無水物に変換するところまでは行わず、ケタールラクトンの構築をやってみた。PMB (p-methoxybenzyl) 基の脱保護はMe3SiSiMe3とI2でMe3SiIを生成させて行ったところ目的物 (2-2) が直接得られた。
 ここまでの研究ではEvansのアルドール反応で (1-1) のC12位の立体化学が制御されることに化合物全体の立体化学が立脚していた。もしこれが何らかの原因で完全に反転しているとしたら面倒なことになると思い、絶対配置を確実なものにするための実験をすることにした。
ここまでの研究ではEvansのアルドール反応で (1-1) のC12位の立体化学が制御されることに化合物全体の立体化学が立脚していた。もしこれが何らかの原因で完全に反転しているとしたら面倒なことになると思い、絶対配置を確実なものにするための実験をすることにした。
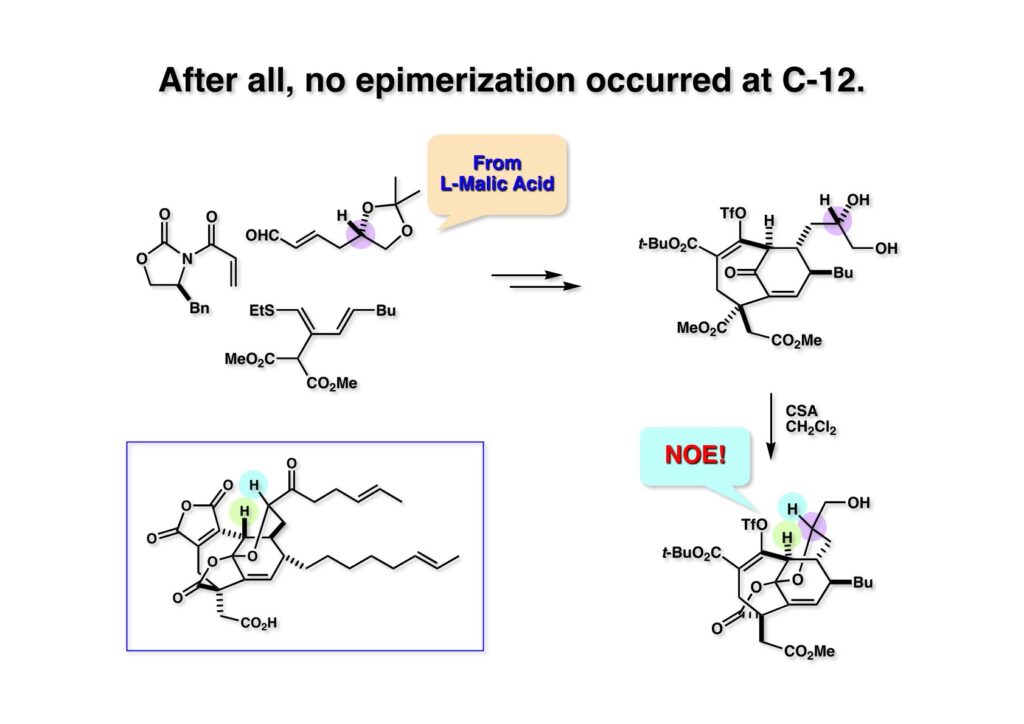 ここではL-リンゴ酸から合成したα,β-不飽和アルデヒド (1-1) を用いてEvansアルドール反応を行って (1-2) に変換し、酸処理することでケタールラクトン (2-2) を得た。この化合物のNOE実験で黄緑と紫でマークしたプロトン間にNOEが観測できたことから (2-2) の絶対配置を決定することができた。
ここではL-リンゴ酸から合成したα,β-不飽和アルデヒド (1-1) を用いてEvansアルドール反応を行って (1-2) に変換し、酸処理することでケタールラクトン (2-2) を得た。この化合物のNOE実験で黄緑と紫でマークしたプロトン間にNOEが観測できたことから (2-2) の絶対配置を決定することができた。
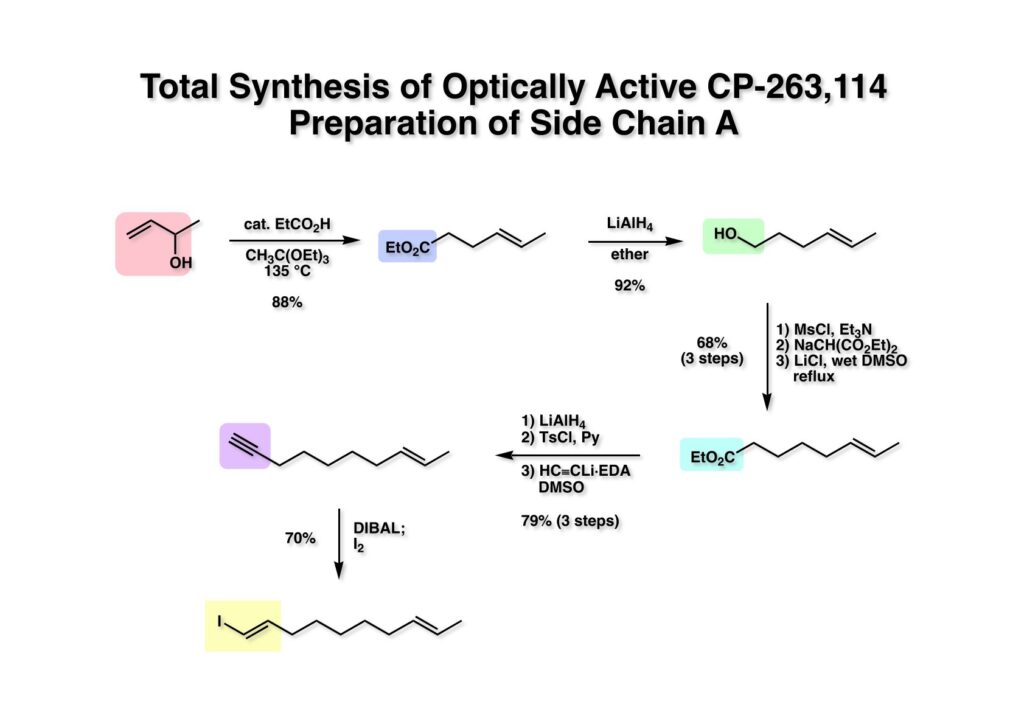 全合成の準備が整ったので側鎖の合成に取りかかった。まず、市販のアルコール (1-1) をIreland-Claisen転位反応でtrans-オレフィン (1-2) に変換した。このエステルをLAH還元してアルコール (1-3) にし、さらに2炭素増炭のため3段階だが確実で大量合成可能な方法を採用した。(1-3) をメシレートにしてからマロン酸エステル塩と反応させ、Krapchoの条件でCO2Me基を除去して (2-2) を得た。(2-2) のLAH還元とトシル化、続く市販のアセチリドとの反応で末端アセチレン体 (2-1) が得られた。これをヒドロアルミニウム化とヨウ素処理でE体のオレフィンに変換した。しかし、今から考えると安価な1,5-dibromopentaneとリチウムアセチリドを組み合わせて合成した方が良かったかもね。
全合成の準備が整ったので側鎖の合成に取りかかった。まず、市販のアルコール (1-1) をIreland-Claisen転位反応でtrans-オレフィン (1-2) に変換した。このエステルをLAH還元してアルコール (1-3) にし、さらに2炭素増炭のため3段階だが確実で大量合成可能な方法を採用した。(1-3) をメシレートにしてからマロン酸エステル塩と反応させ、Krapchoの条件でCO2Me基を除去して (2-2) を得た。(2-2) のLAH還元とトシル化、続く市販のアセチリドとの反応で末端アセチレン体 (2-1) が得られた。これをヒドロアルミニウム化とヨウ素処理でE体のオレフィンに変換した。しかし、今から考えると安価な1,5-dibromopentaneとリチウムアセチリドを組み合わせて合成した方が良かったかもね。
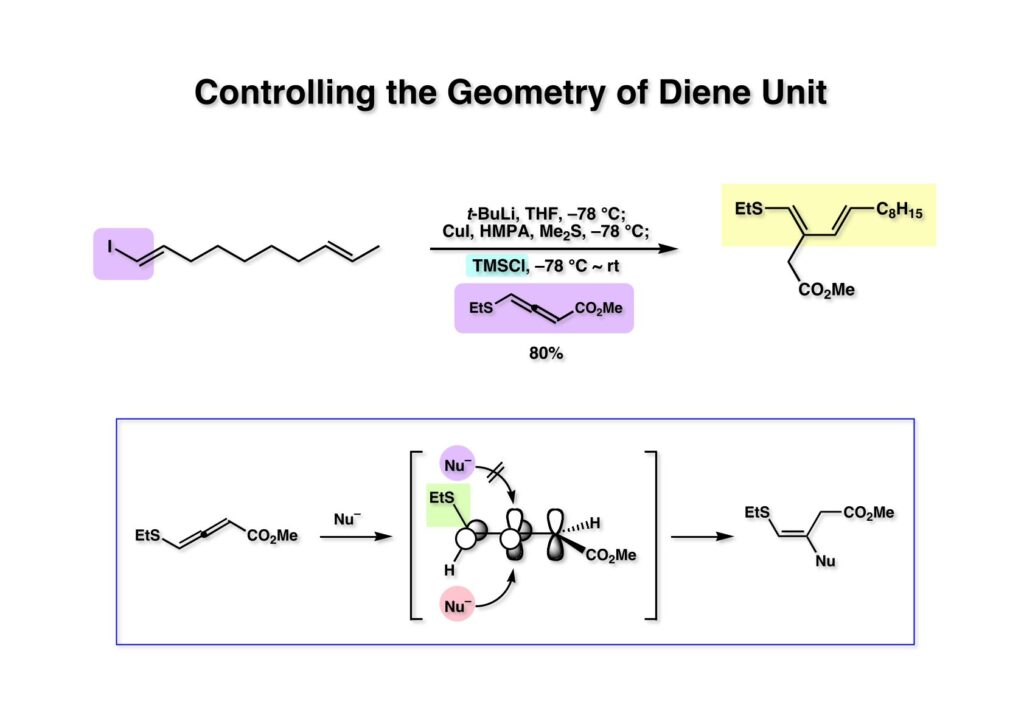 (1-1) を桑嶋研が報告した方法でアルケニル銅をアレニルエステル (1-2) に共役付加させてジエン体 (1-3) を得た。下のスキームはアレンへの付加で立体化学を制御する常道を示している。
(1-1) を桑嶋研が報告した方法でアルケニル銅をアレニルエステル (1-2) に共役付加させてジエン体 (1-3) を得た。下のスキームはアレンへの付加で立体化学を制御する常道を示している。
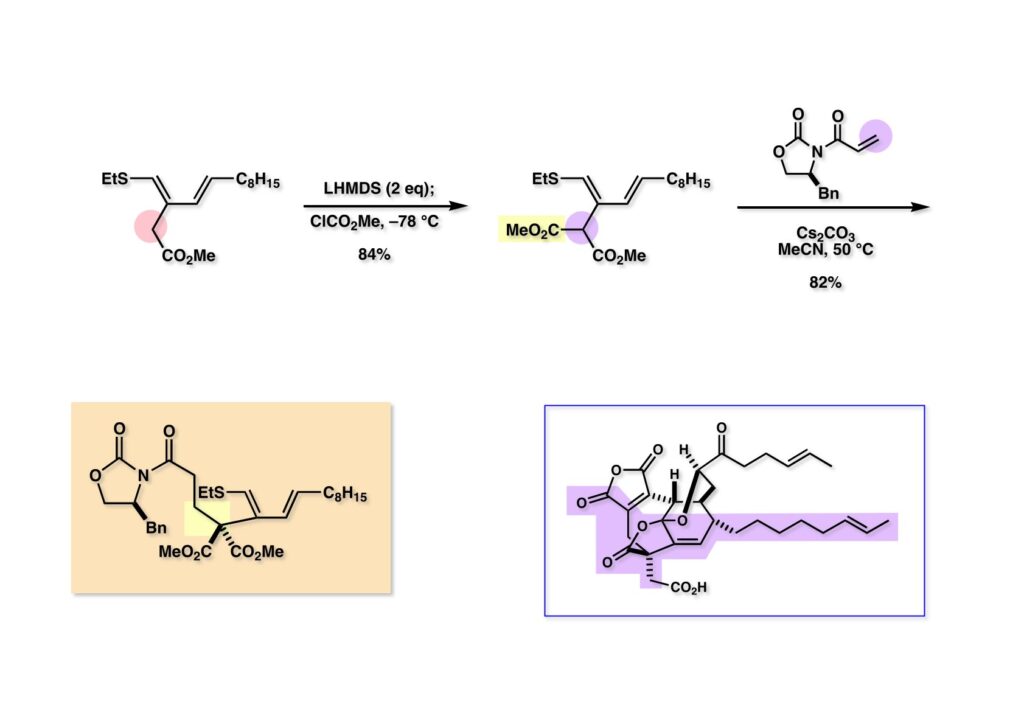 前述のモデル化合物で説明した経路で (1-1) から (2-1) へ変換した。次は上の側鎖を合成する番である。
前述のモデル化合物で説明した経路で (1-1) から (2-1) へ変換した。次は上の側鎖を合成する番である。
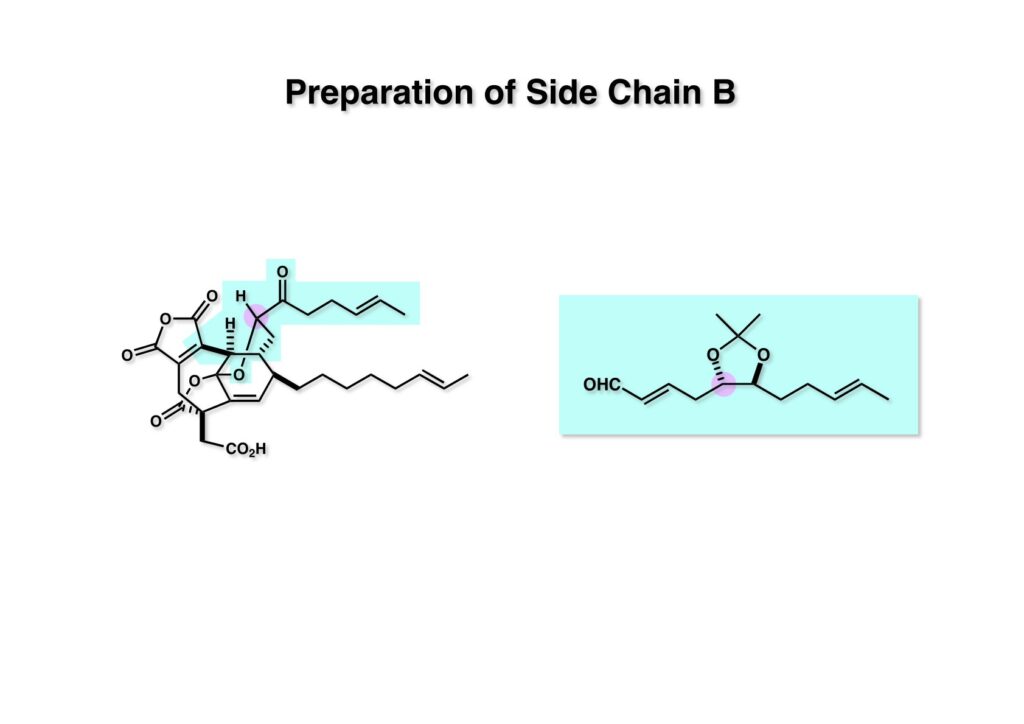 上の側鎖部分は (1-2) のような光学活性α,β-不飽和アルデヒドを合成することにした。
上の側鎖部分は (1-2) のような光学活性α,β-不飽和アルデヒドを合成することにした。
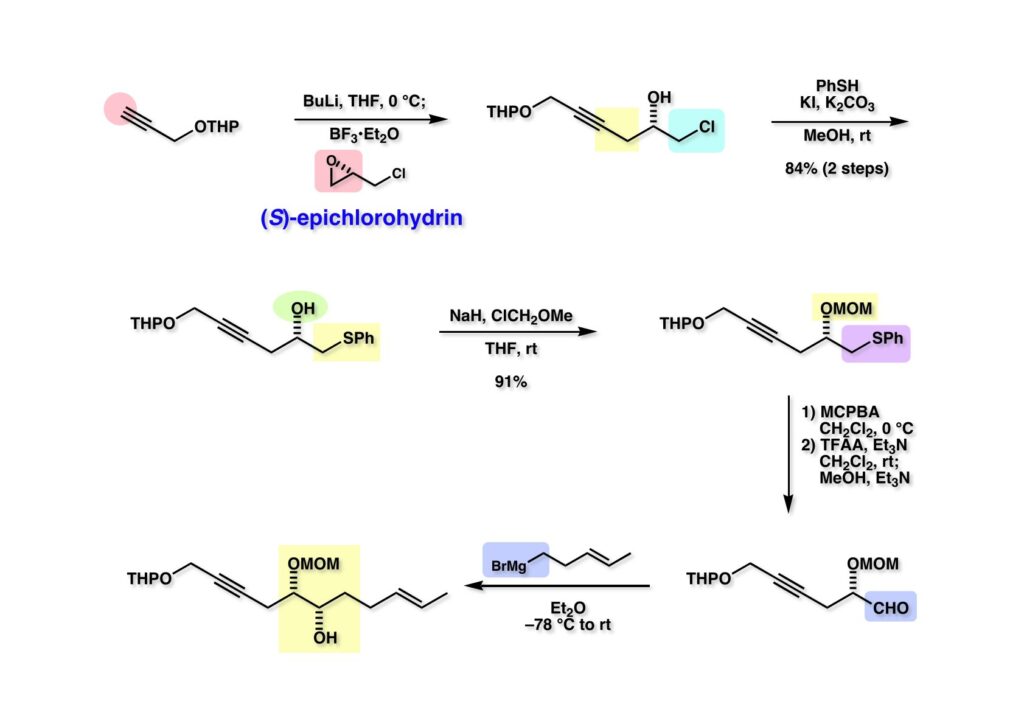 まず市販のプロパルギルアルコールをTHPエーテル (1-1) として保護し、次にBuLIで脱プロトン化後にBF3·Et2O存在下で(S)-epichlorohydrinと反応させるとエポキサイドが開裂して (1-2) が高収率で得られた。末端の炭素をアルデヒドに変換したいのでクロライドをチオフェノールで置換して (2-1) を得た。アルデヒドへのGrignard付加ではキレーション制御をしたいので (2-1) の水酸基をMOM基で保護して (2-2) を得た。フェニルサルファイドのPummerer転位は問題なく進行した。前に述べたように無水トリフロロ酢酸を用いるのが一番容易にPummerer転位が進行するし、反応終了後にMeOHとEt3Nを加えるだけでアルデヒド (3-3) が得られて簡便な方法である。このアルデヒドにGrignard試薬 (3-2) を低温で付加させると予想通りキレーション制御で単一の付加体 (3-1) が得られた。生じた水酸基は後にケトンになるところだが単一物が得られるに越したことはない。
まず市販のプロパルギルアルコールをTHPエーテル (1-1) として保護し、次にBuLIで脱プロトン化後にBF3·Et2O存在下で(S)-epichlorohydrinと反応させるとエポキサイドが開裂して (1-2) が高収率で得られた。末端の炭素をアルデヒドに変換したいのでクロライドをチオフェノールで置換して (2-1) を得た。アルデヒドへのGrignard付加ではキレーション制御をしたいので (2-1) の水酸基をMOM基で保護して (2-2) を得た。フェニルサルファイドのPummerer転位は問題なく進行した。前に述べたように無水トリフロロ酢酸を用いるのが一番容易にPummerer転位が進行するし、反応終了後にMeOHとEt3Nを加えるだけでアルデヒド (3-3) が得られて簡便な方法である。このアルデヒドにGrignard試薬 (3-2) を低温で付加させると予想通りキレーション制御で単一の付加体 (3-1) が得られた。生じた水酸基は後にケトンになるところだが単一物が得られるに越したことはない。
 (1-1) のMOM基とTHP基をメタノール中CSAで加メタノール分解してトリオール (1-2) を得た。CSA-CuSO4-アセトン環流という条件であればジオールがアセトナイドになるだけだが時間がかかるのでアセトンジメチルケタールとCSAで処理してアセトナイドとアセトンの混合ケタールに変換してからメタノール中PPTSで混合ケタールを外して (2-2) を得た。次にLiAlH4で処理すると分子内ヒドロアルミニウム化が進行してtrans-アリルアルコールが得られた。これをSwern酸化するとα,β-不飽和アルデヒド (2-1) が得られる。
(1-1) のMOM基とTHP基をメタノール中CSAで加メタノール分解してトリオール (1-2) を得た。CSA-CuSO4-アセトン環流という条件であればジオールがアセトナイドになるだけだが時間がかかるのでアセトンジメチルケタールとCSAで処理してアセトナイドとアセトンの混合ケタールに変換してからメタノール中PPTSで混合ケタールを外して (2-2) を得た。次にLiAlH4で処理すると分子内ヒドロアルミニウム化が進行してtrans-アリルアルコールが得られた。これをSwern酸化するとα,β-不飽和アルデヒド (2-1) が得られる。
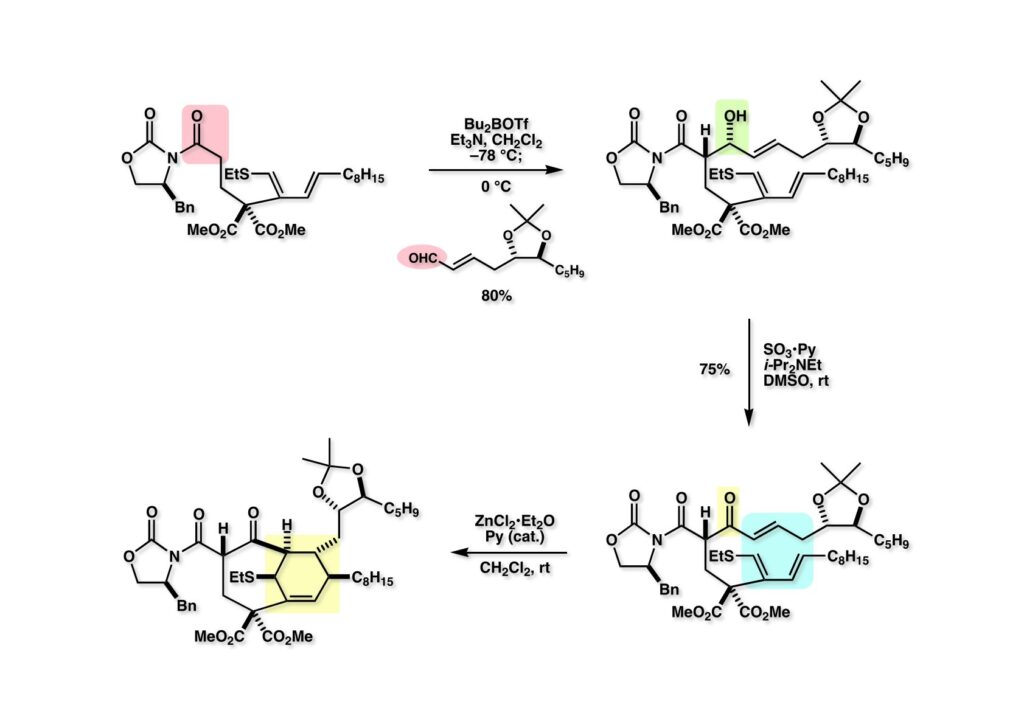 モデル化合物同様、前ページで得られたα,β-不飽和アルデヒド (1-2) と (1-1) のEvansアルドール反応は (1-3) を高収率で与えた。これをParikh-Doering酸化して得られたケトン (2-2) を塩化亜鉛で処理すると分子内Diels-Alder反応が進行して目的物 (2-1) のみが得られた。
モデル化合物同様、前ページで得られたα,β-不飽和アルデヒド (1-2) と (1-1) のEvansアルドール反応は (1-3) を高収率で与えた。これをParikh-Doering酸化して得られたケトン (2-2) を塩化亜鉛で処理すると分子内Diels-Alder反応が進行して目的物 (2-1) のみが得られた。
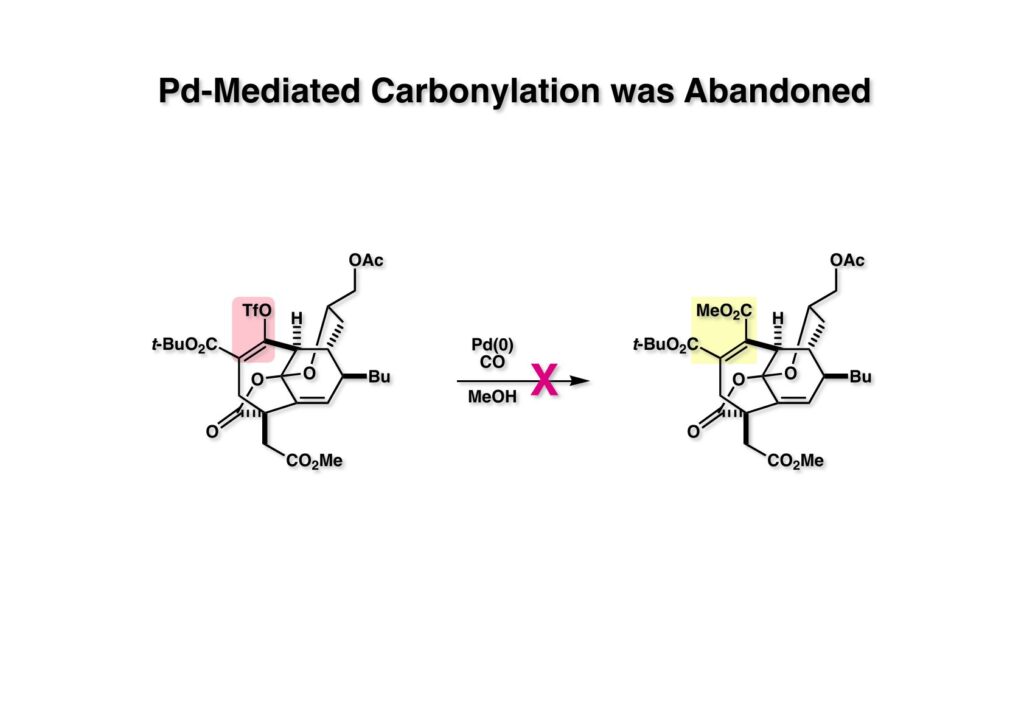 モデル実験でケタールラクトン (1-1) に変換してからエノールトリフレートのカルボニル化を試したが全く進行しなかった。本来ならばPummerer転位後のケトンの段階でPd触媒を用いたカルボニル化の条件をもっと検討すべきだったと思う。この反省を後の2度目の全合成には生かしている。
モデル実験でケタールラクトン (1-1) に変換してからエノールトリフレートのカルボニル化を試したが全く進行しなかった。本来ならばPummerer転位後のケトンの段階でPd触媒を用いたカルボニル化の条件をもっと検討すべきだったと思う。この反省を後の2度目の全合成には生かしている。
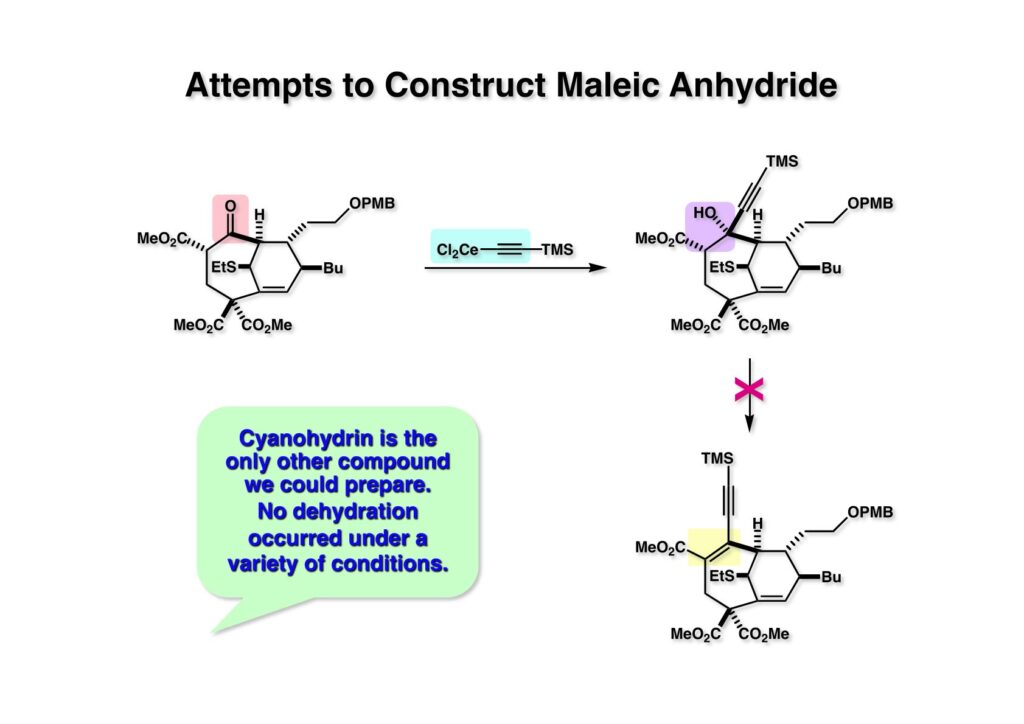 β-ケトエステルからマレイン酸無水物を構築するのはかなり色々な方法を使った。このβ-ケトエステルは平面上に並びにくいためにエノール化しにくく、ケトンとして独立に反応した。シアンヒドリンやアセチリドを付加した化合物から水酸基の脱離を試みたがうまくいかなかった。
β-ケトエステルからマレイン酸無水物を構築するのはかなり色々な方法を使った。このβ-ケトエステルは平面上に並びにくいためにエノール化しにくく、ケトンとして独立に反応した。シアンヒドリンやアセチリドを付加した化合物から水酸基の脱離を試みたがうまくいかなかった。
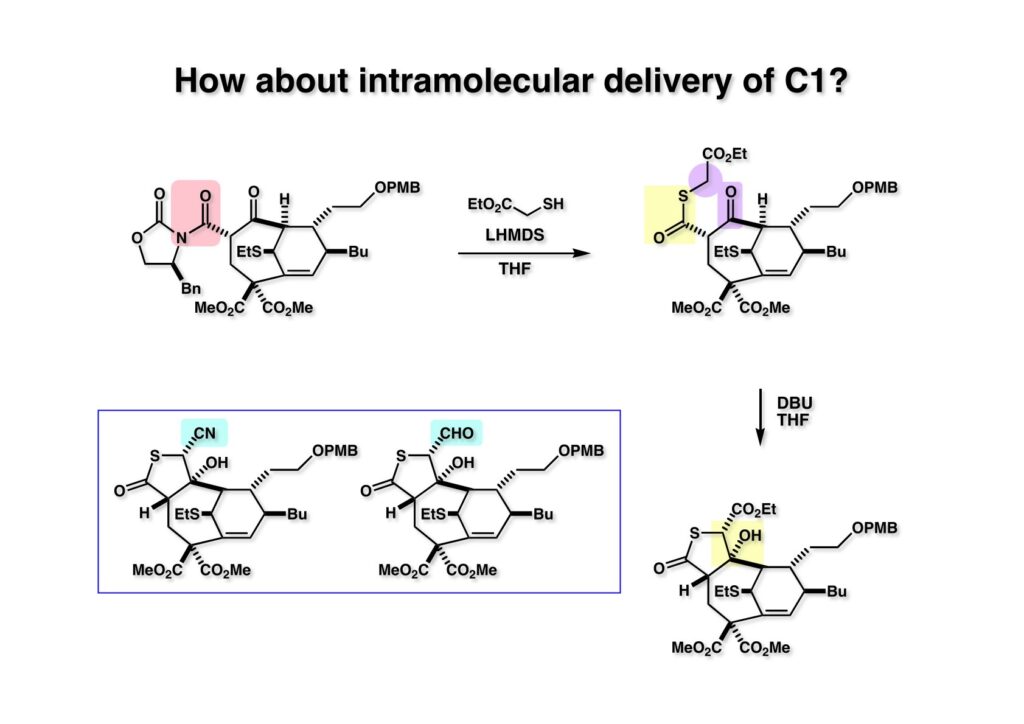 Evansの不斉補助基はチオレートで除去できるので (1-1) を3種類のチオレートで処理し、得られたチオエステルをDBUで環化すると (2-1)、(2-2)、(2-3) が容易に得られた。
Evansの不斉補助基はチオレートで除去できるので (1-1) を3種類のチオレートで処理し、得られたチオエステルをDBUで環化すると (2-1)、(2-2)、(2-3) が容易に得られた。
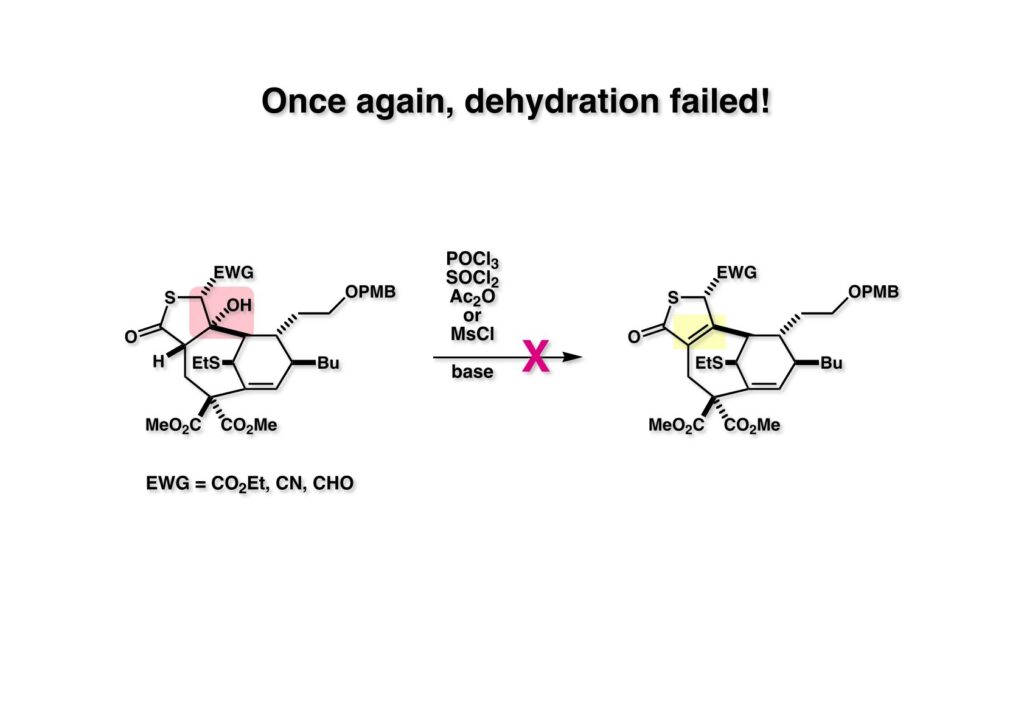 ところが、又しても (1-1) の水酸基が脱離せず、(1-2) が得られなかった。これは極めて大きな立体障害によるとしか考えられないが…。
ところが、又しても (1-1) の水酸基が脱離せず、(1-2) が得られなかった。これは極めて大きな立体障害によるとしか考えられないが…。
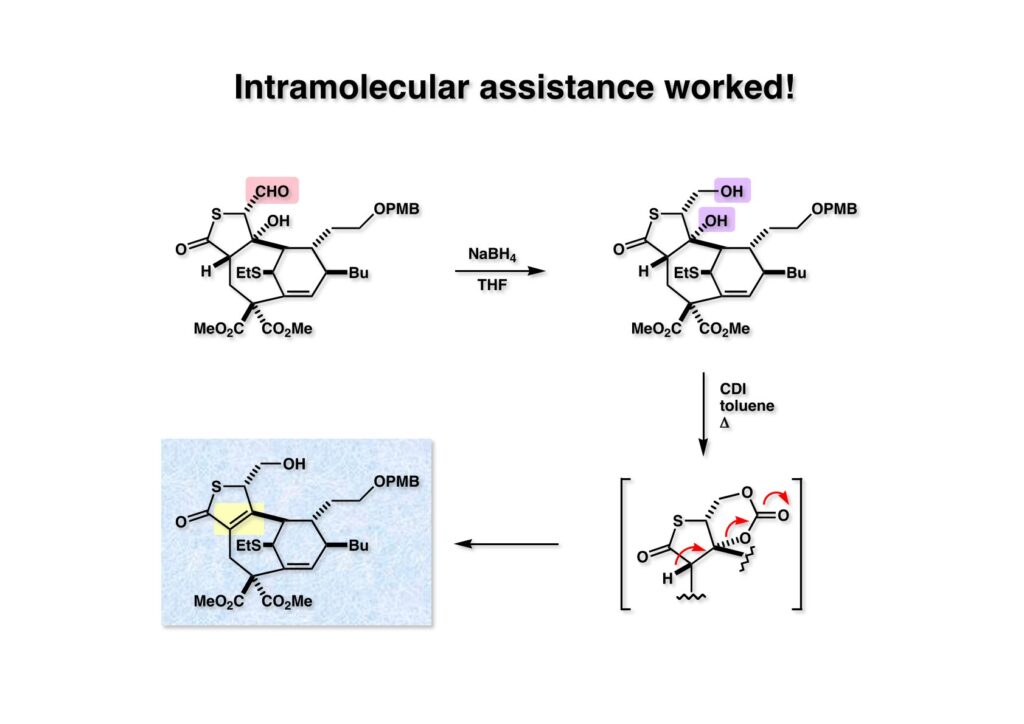 そこで (1-1) の水酸基が立体障害によって試薬が届かないかもしれないので分子内から活性化しようと考えた。アルデヒド (1-1) を還元してジオール体 (1-2) とし、これにcarbonyl diimidazole (CDI) を作用させて環状カーボネート (2-2) を構築しようとしたところ脱離が進行してチオブテノライド (2-1) が得られた。
そこで (1-1) の水酸基が立体障害によって試薬が届かないかもしれないので分子内から活性化しようと考えた。アルデヒド (1-1) を還元してジオール体 (1-2) とし、これにcarbonyl diimidazole (CDI) を作用させて環状カーボネート (2-2) を構築しようとしたところ脱離が進行してチオブテノライド (2-1) が得られた。
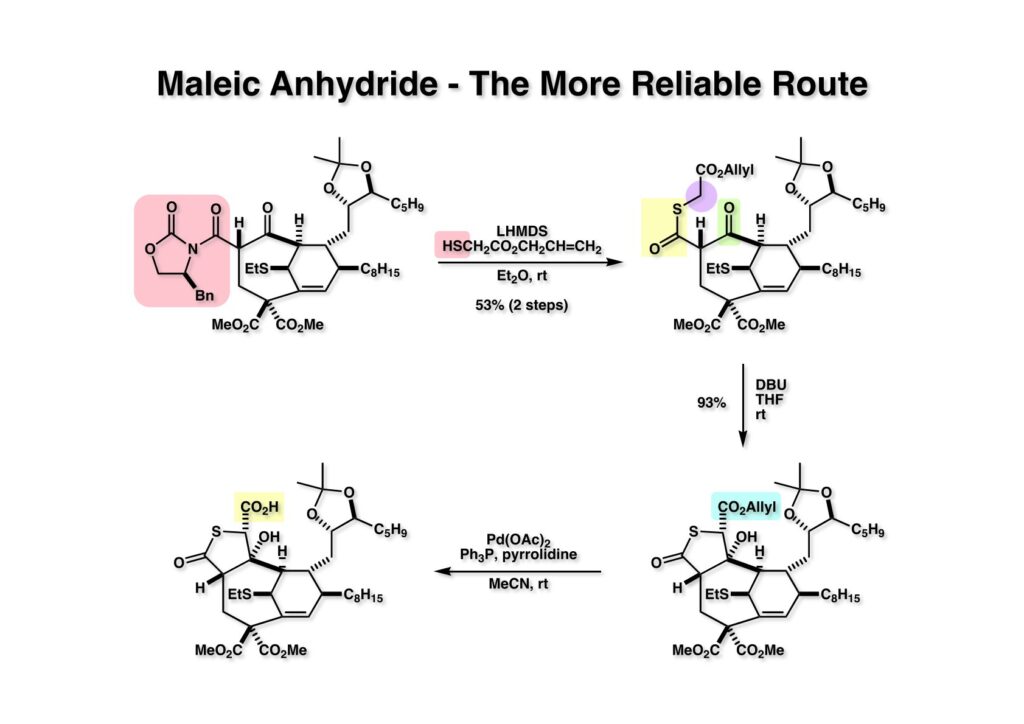 そこで、今度はβ-ラクトンを構築してみようと考えた。β-ラクトンは加熱するとオレフィンが得られることは広く知られている。(1-1) をallyl mercaptoacetate (1-2) とLHMDSでチオエステル (1-3) に変換し、DBUで感化させると (2-2) が得られた。53%の収率で2段階とあるのは分子内Diels-Alder反応を含めている。(2-2) のアリスエステルをPd触媒で除いてカルボン酸 (2-1) を得た。
そこで、今度はβ-ラクトンを構築してみようと考えた。β-ラクトンは加熱するとオレフィンが得られることは広く知られている。(1-1) をallyl mercaptoacetate (1-2) とLHMDSでチオエステル (1-3) に変換し、DBUで感化させると (2-2) が得られた。53%の収率で2段階とあるのは分子内Diels-Alder反応を含めている。(2-2) のアリスエステルをPd触媒で除いてカルボン酸 (2-1) を得た。
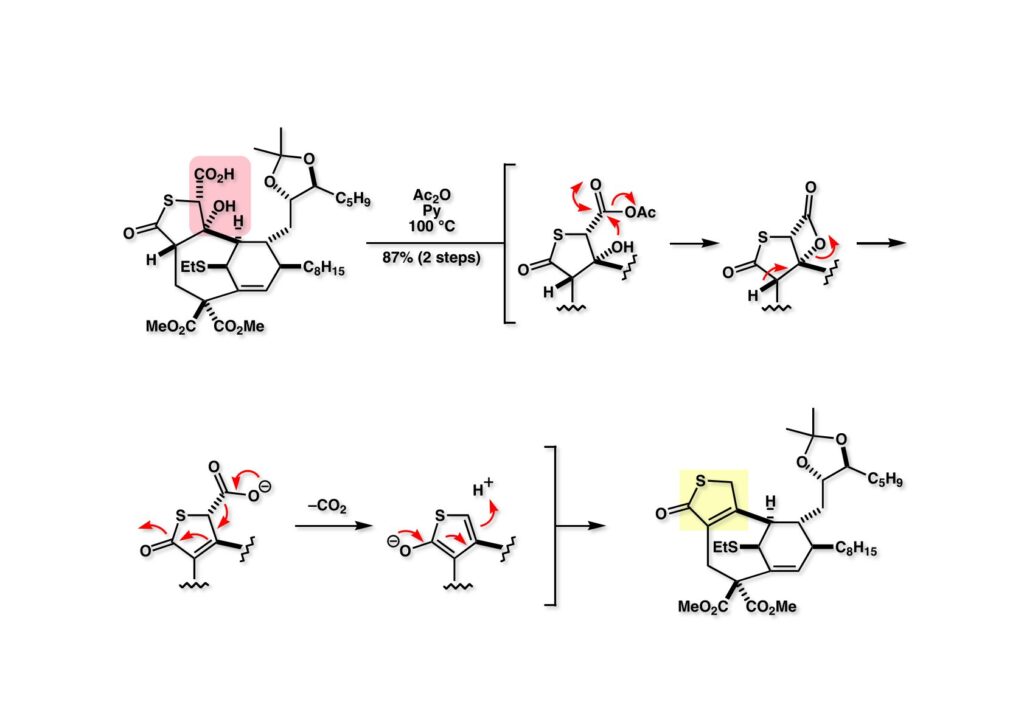 (1-1) を無水酢酸-Pyで加熱したところ、おそらくβ-ラクトン (1-3) 経由で脱炭酸が進行してチオブテノライド (2-3) が高収率で得られた。
(1-1) を無水酢酸-Pyで加熱したところ、おそらくβ-ラクトン (1-3) 経由で脱炭酸が進行してチオブテノライド (2-3) が高収率で得られた。
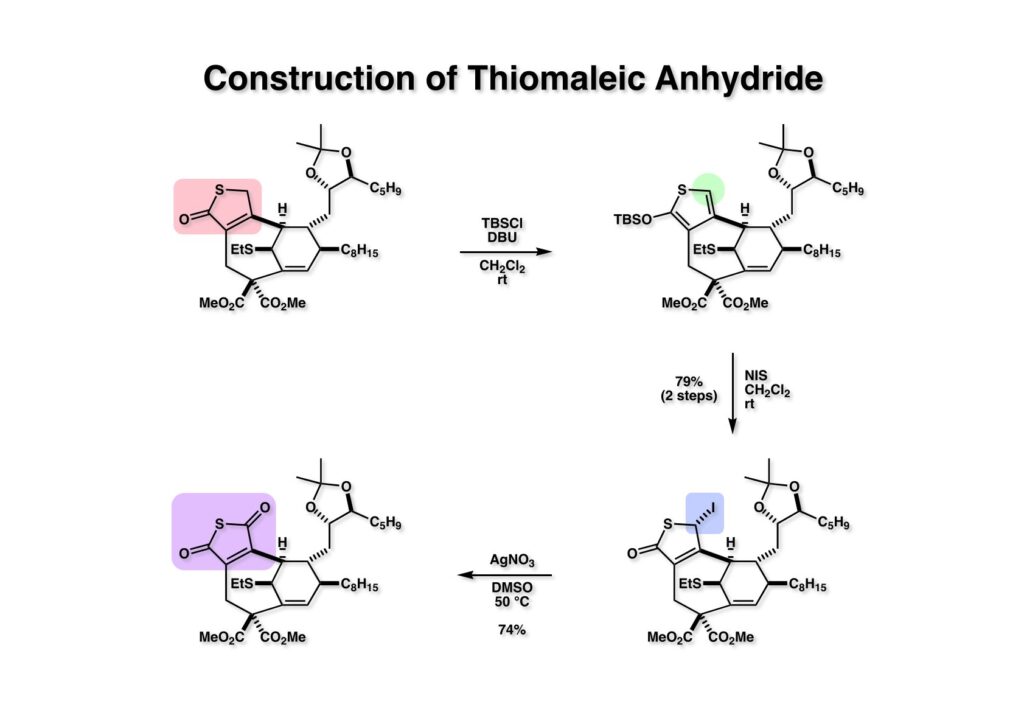 チオブテノライド (1-1) をチオマレイン酸無水物 (2-1) に変換するにはチオフェン誘導体を経由することにした。先ず、(1-1) をTBSClとDBUを用いてチオフェン誘導体 (1-2) にし、NIS (N-iodosuccinimide) を用いて5位をヨウ素化して (2-2) を得た。ここで思い出したのはPhCH2BrをNaHCO3存在下でDMSO中で加熱するとPhCHOに変換する反応だ。人名反応だと思うが名前を忘れてしまった。(2-1) をDMSO中で硝酸銀と加熱すると期待どおりチオマレイン酸無水物 (2-1) が生成した。Swern酸化の中間体をイメージしてもらえば良い。
チオブテノライド (1-1) をチオマレイン酸無水物 (2-1) に変換するにはチオフェン誘導体を経由することにした。先ず、(1-1) をTBSClとDBUを用いてチオフェン誘導体 (1-2) にし、NIS (N-iodosuccinimide) を用いて5位をヨウ素化して (2-2) を得た。ここで思い出したのはPhCH2BrをNaHCO3存在下でDMSO中で加熱するとPhCHOに変換する反応だ。人名反応だと思うが名前を忘れてしまった。(2-1) をDMSO中で硝酸銀と加熱すると期待どおりチオマレイン酸無水物 (2-1) が生成した。Swern酸化の中間体をイメージしてもらえば良い。
 チオマレイン酸無水物はアルカリ条件で簡単に加水分解され、酸性に戻すとすぐにマレイン酸無水物になることは確認した。ここではBa(OH)2でさらに加水分解を続けると、β-配置マロネートのみが加水分解されることが判明した。このカルボン酸は1炭素増炭すべきものなので、得られた (1-2) を塩化オキザリルで酸クロライドに変換してからジアゾメタンと反応させてジアゾケトン (2-2) を得た。次に (2-2) を銀塩 (PhCO2Ag) とt-BuOH中で加熱すると有名なArndt-Eistert反応が進行して (2-1) が得られた。
チオマレイン酸無水物はアルカリ条件で簡単に加水分解され、酸性に戻すとすぐにマレイン酸無水物になることは確認した。ここではBa(OH)2でさらに加水分解を続けると、β-配置マロネートのみが加水分解されることが判明した。このカルボン酸は1炭素増炭すべきものなので、得られた (1-2) を塩化オキザリルで酸クロライドに変換してからジアゾメタンと反応させてジアゾケトン (2-2) を得た。次に (2-2) を銀塩 (PhCO2Ag) とt-BuOH中で加熱すると有名なArndt-Eistert反応が進行して (2-1) が得られた。
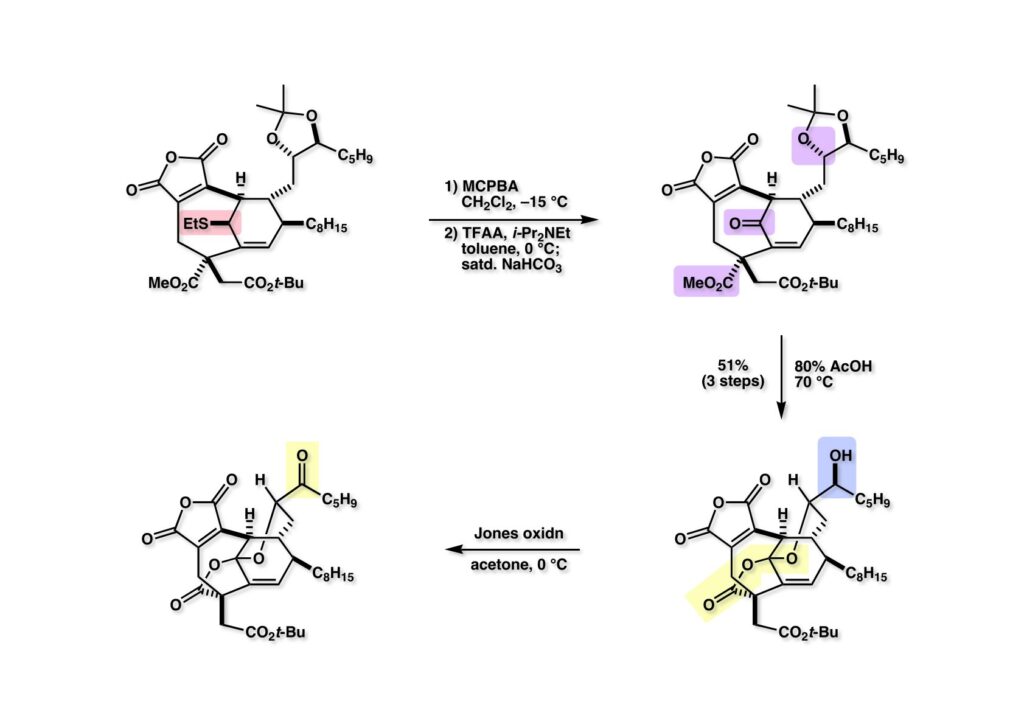 (1-1) を1当量のMCPBAでスルホキサイドにし、TFAAとHünig baseで処理するとPummerer転位が起こり、重曹水を加えて後処理するとケトン (1-2) が得られた。アセトナイド (1-2) を含水酢酸中で加水分解してケタールラクトン (2-2) を得た後に水酸基をJones酸化することでケトン (2-1) が得られた。
(1-1) を1当量のMCPBAでスルホキサイドにし、TFAAとHünig baseで処理するとPummerer転位が起こり、重曹水を加えて後処理するとケトン (1-2) が得られた。アセトナイド (1-2) を含水酢酸中で加水分解してケタールラクトン (2-2) を得た後に水酸基をJones酸化することでケトン (2-1) が得られた。
 (1-1) をギ酸で室温放置するとt-butylエステルが外れてCP-263,114が得られた。旋光度を測定したところ、ほぼ天然物と一致したため天然物そのものの全合成に成功したと言える。K. C. NicolaouやMatt Shairは光学異性体を合成してしまった。まあ、安いL-phenylalanineを使ったので偶然天然物に辿り着いただけだけど。ところで、Matt Shairと我々の全合成はJACSのオフィスに同時に到着し、エディターはback-to-backでパブリッシュすると言っていたのに、Mattの論文の方が先に出たのは納得できない。まあ、別にどうだっていいけど、エディター(誰だとは言わないけど)も結構いい加減なものだ。
(1-1) をギ酸で室温放置するとt-butylエステルが外れてCP-263,114が得られた。旋光度を測定したところ、ほぼ天然物と一致したため天然物そのものの全合成に成功したと言える。K. C. NicolaouやMatt Shairは光学異性体を合成してしまった。まあ、安いL-phenylalanineを使ったので偶然天然物に辿り着いただけだけど。ところで、Matt Shairと我々の全合成はJACSのオフィスに同時に到着し、エディターはback-to-backでパブリッシュすると言っていたのに、Mattの論文の方が先に出たのは納得できない。まあ、別にどうだっていいけど、エディター(誰だとは言わないけど)も結構いい加減なものだ。
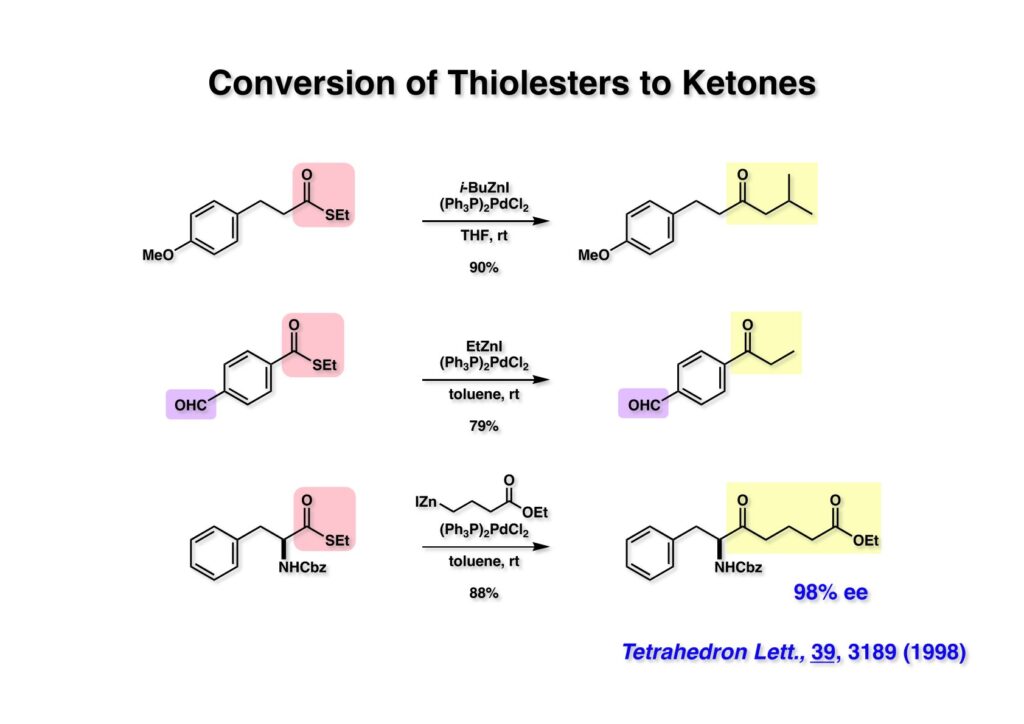 ライス大学でチオエステルをアルデヒドに変換する新規反応を開発したが、PdがC-S結合に酸化的付加をしていることは確実なので助手の徳山英利さんにケトン合成の開発を頼んだ。すぐに根岸カップリングの条件が適用できることが分かった。この反応の面白いところは (2-1) のようなアルデヒドが共存しているチオエステルもアルデヒドを保護することなくケトンに変換できることである。CP-263,114には無水マレイン酸という非常に反応性の高い官能基があるが、それでもチオエステルをケトンに変換できることを示そうと2nd generationの全合成を開始した。
ライス大学でチオエステルをアルデヒドに変換する新規反応を開発したが、PdがC-S結合に酸化的付加をしていることは確実なので助手の徳山英利さんにケトン合成の開発を頼んだ。すぐに根岸カップリングの条件が適用できることが分かった。この反応の面白いところは (2-1) のようなアルデヒドが共存しているチオエステルもアルデヒドを保護することなくケトンに変換できることである。CP-263,114には無水マレイン酸という非常に反応性の高い官能基があるが、それでもチオエステルをケトンに変換できることを示そうと2nd generationの全合成を開始した。
 この反応経路は1st generation全合成と同様なので説明は省略する。
この反応経路は1st generation全合成と同様なので説明は省略する。
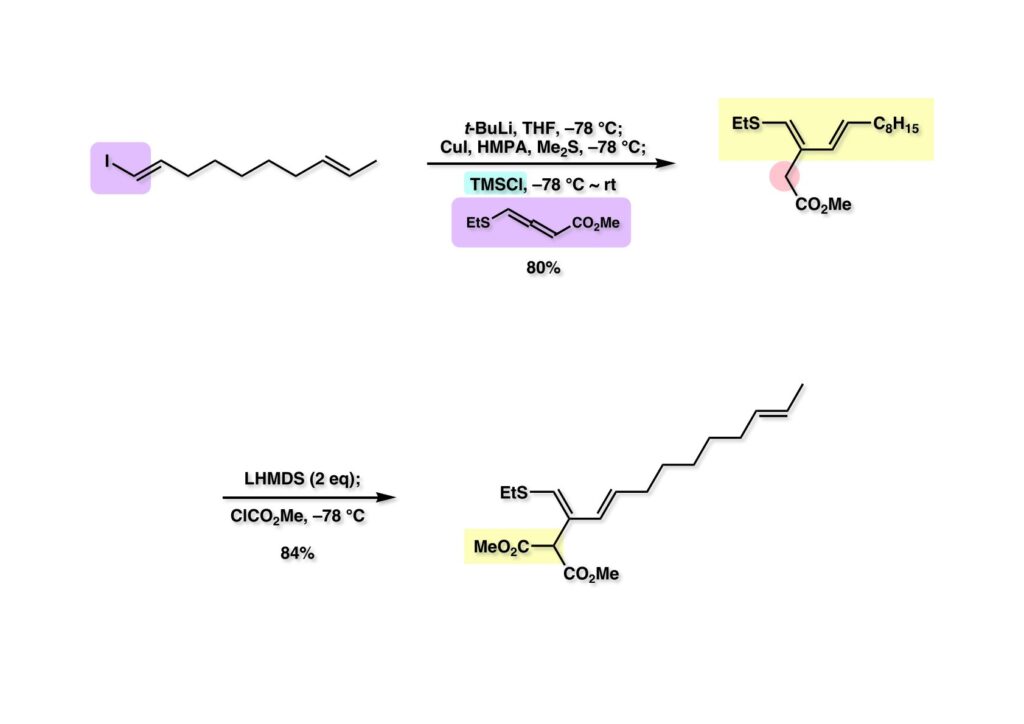 このマロネート (2-1) も1st generation全合成と同じであるので、さかのぼって思い出してほしい。
このマロネート (2-1) も1st generation全合成と同じであるので、さかのぼって思い出してほしい。
 L-Malic acid (リンゴ酸) から合成したα,β-不飽和アルデヒド (2-1) を用いて (3-1) を合成する経路も1st generation全合成と同様で、特にコメントは必要ない。
L-Malic acid (リンゴ酸) から合成したα,β-不飽和アルデヒド (2-1) を用いて (3-1) を合成する経路も1st generation全合成と同様で、特にコメントは必要ない。
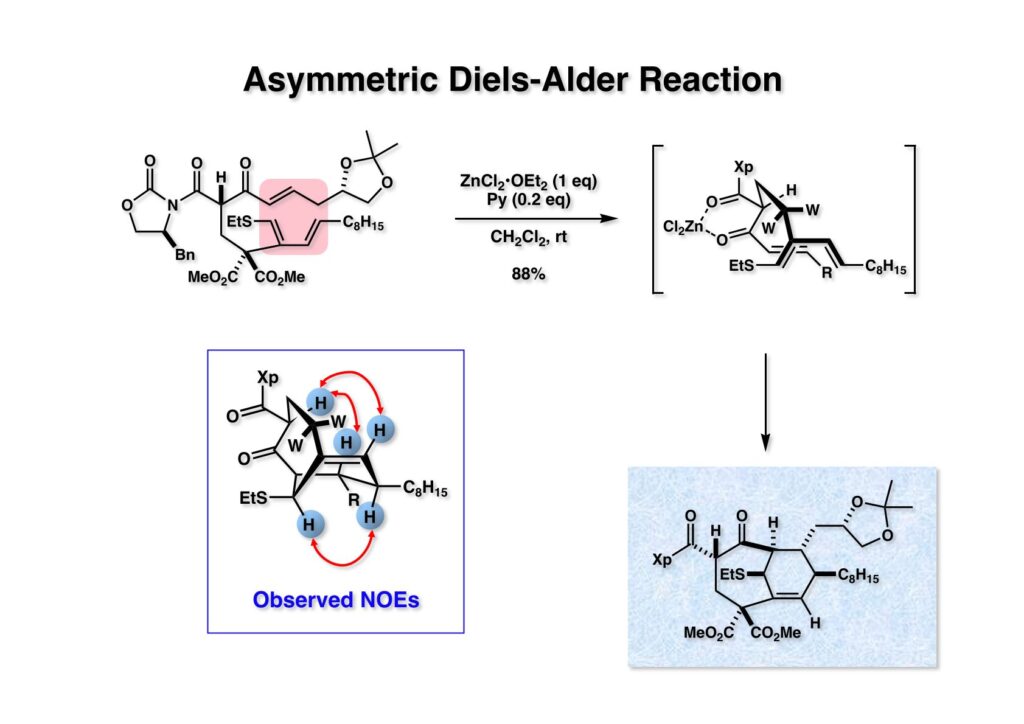 (1-1) の分子内Diels-Alderも収率良く進行し、単一の生成物 (2-2) を与えた。
(1-1) の分子内Diels-Alderも収率良く進行し、単一の生成物 (2-2) を与えた。
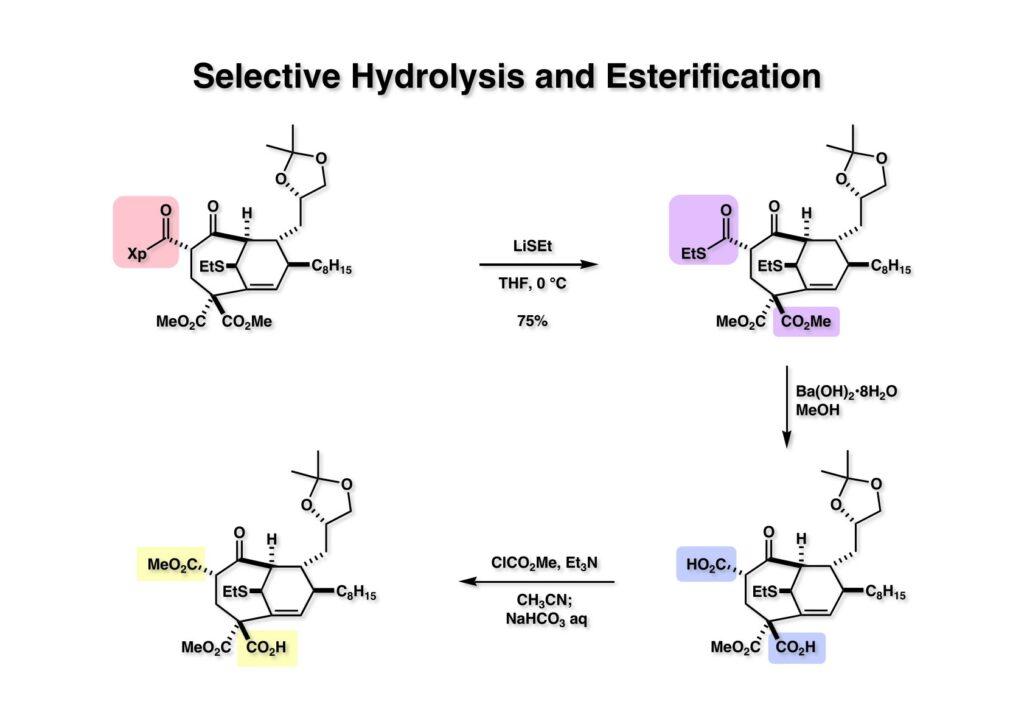 (1-1) のEvans不斉補助基をLiSEtで処理してチオエステル (1-2) に変換し、さらにメタノール中でBα(OH)2で加水分解を試みると、ジカルボン酸 (2-2) が得られた。これをClCO2Me-Et3Nで処理すると上のカルボン酸はメチルエステルに、下のカルボン酸は混合酸無水物になる。これに重曹水を加えると混合酸無水物は加水分解されて (2-1) が得られた。β-ケトカルボン酸がこの条件でメチルエステルに変換される反応機構は混合酸無水物の生成後にメタノールが脱離して反応性が大きいケテンが生成し、これにメタノールが付加すると考えている。
(1-1) のEvans不斉補助基をLiSEtで処理してチオエステル (1-2) に変換し、さらにメタノール中でBα(OH)2で加水分解を試みると、ジカルボン酸 (2-2) が得られた。これをClCO2Me-Et3Nで処理すると上のカルボン酸はメチルエステルに、下のカルボン酸は混合酸無水物になる。これに重曹水を加えると混合酸無水物は加水分解されて (2-1) が得られた。β-ケトカルボン酸がこの条件でメチルエステルに変換される反応機構は混合酸無水物の生成後にメタノールが脱離して反応性が大きいケテンが生成し、これにメタノールが付加すると考えている。
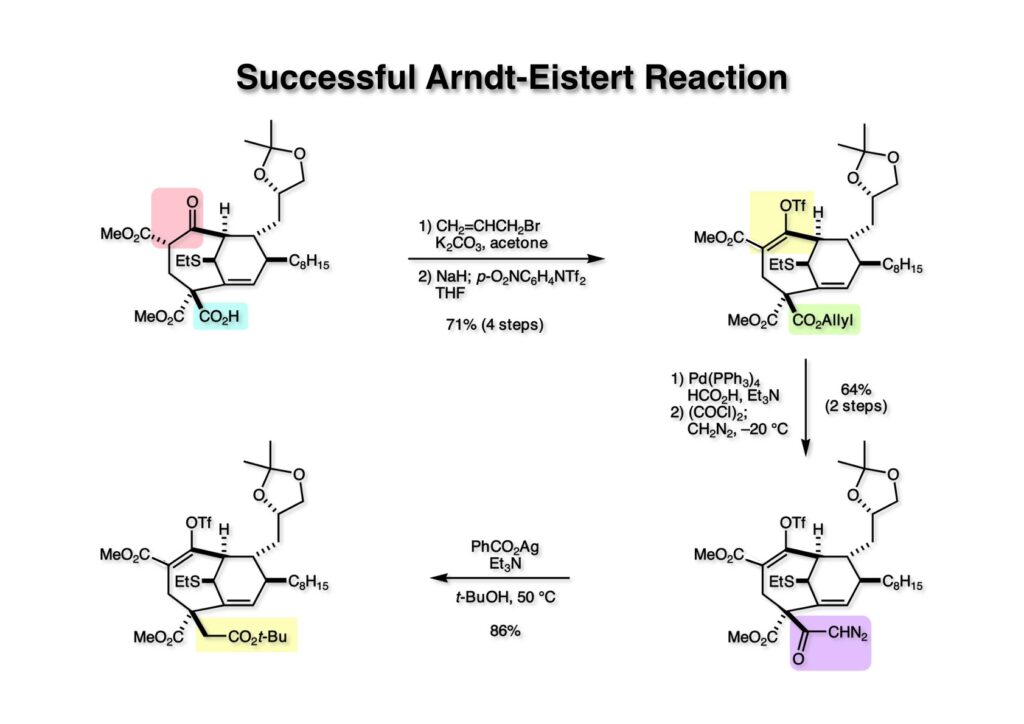 (1-1) のカルボン酸を後で選択的に戻すためにアリルエステルにしてからβ-ケトエステルをエノールトリフレート (1-2) に変換した。次にアリルエステルをPd触媒を使ってカルボン酸にし、酸クロライド経由でジアゾケトン (2-2) を得た。PhCO2Agを触媒にしてArndt-Eistert反応をt-BuOH中で行って (2-1) が得られた。
(1-1) のカルボン酸を後で選択的に戻すためにアリルエステルにしてからβ-ケトエステルをエノールトリフレート (1-2) に変換した。次にアリルエステルをPd触媒を使ってカルボン酸にし、酸クロライド経由でジアゾケトン (2-2) を得た。PhCO2Agを触媒にしてArndt-Eistert反応をt-BuOH中で行って (2-1) が得られた。
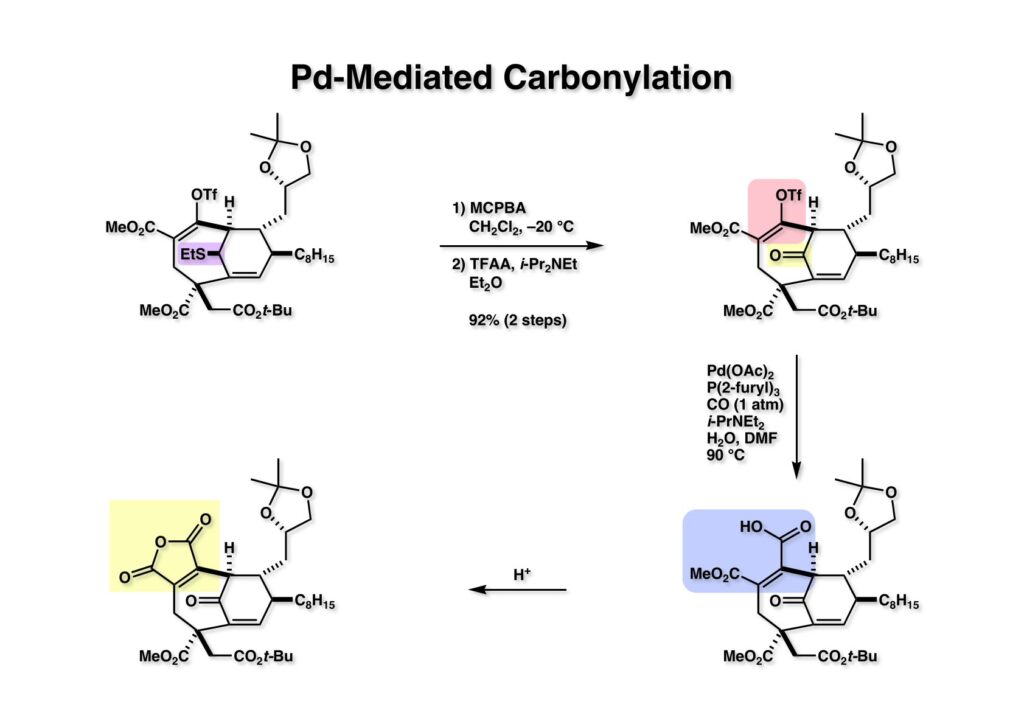 (1-1) をPummerer転位でケトン (1-2) に変換後、Pd触媒を用いたカルボニル化を含水DMFで行ってカルボン酸 (2-2) を得た。これを酸処理すると容易にマレイン酸無水物 (2-1) に変換することができた。
(1-1) をPummerer転位でケトン (1-2) に変換後、Pd触媒を用いたカルボニル化を含水DMFで行ってカルボン酸 (2-2) を得た。これを酸処理すると容易にマレイン酸無水物 (2-1) に変換することができた。
 アセトナイド (1-1) を酢酸中で加熱するとジオールが生成し、直ちにケタールラクトン (1-2) が得られた。(1-2) の一級アルコールをJones酸化することでカルボン酸 (2-2) を得た。これを塩化オキザリルで酸クロライドにしてからEtSHと反応させてチオエステル (2-1) が得られた。
アセトナイド (1-1) を酢酸中で加熱するとジオールが生成し、直ちにケタールラクトン (1-2) が得られた。(1-2) の一級アルコールをJones酸化することでカルボン酸 (2-2) を得た。これを塩化オキザリルで酸クロライドにしてからEtSHと反応させてチオエステル (2-1) が得られた。
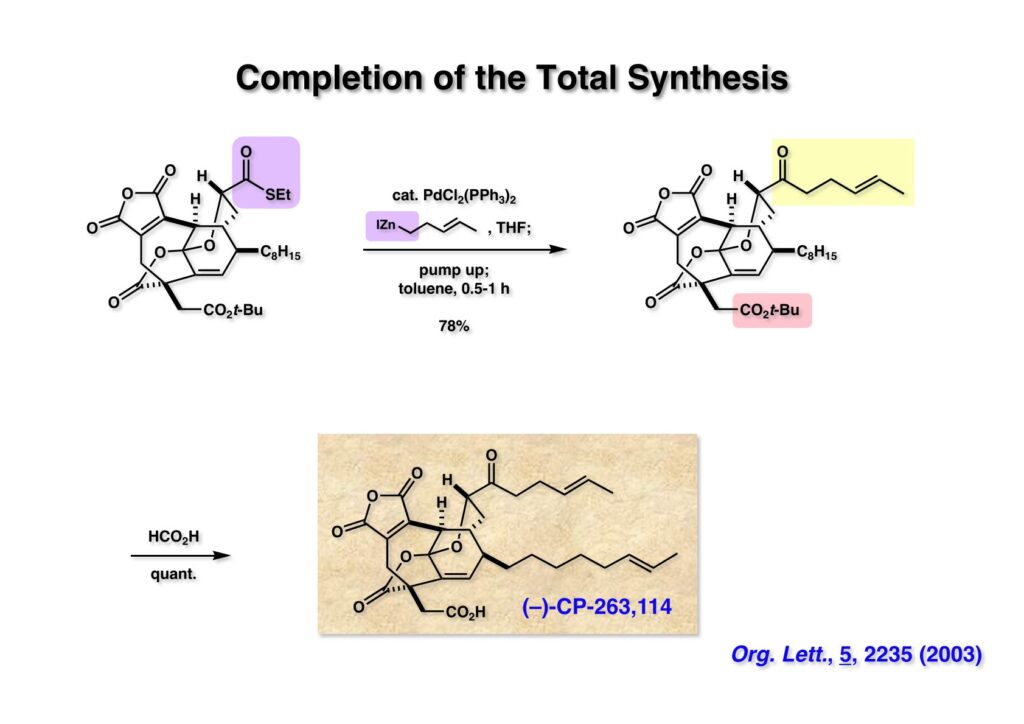 2nd generaton全合成の最大の山場である「福山カップリング」によるチオエステル (1-1) のケトン (1-2) への変換は問題なく進行し、反応性の高い無水マレイン酸共存下でもこのカップリング反応は使えることが証明された。このケトンはエピメリ化しやすく、シリカゲルで精製後にほんの少しだがエピマーが観測された。Danishefskyは確かエピマーの方を合成して、これも天然物だ、と言っていた記憶があるが面倒なので文献はチェックしない。残るはt-ブチルエステルをカルボン酸に変換するだけで、ギ酸中室温放置で定量的に(-)-CP-263,114が得られた。
2nd generaton全合成の最大の山場である「福山カップリング」によるチオエステル (1-1) のケトン (1-2) への変換は問題なく進行し、反応性の高い無水マレイン酸共存下でもこのカップリング反応は使えることが証明された。このケトンはエピメリ化しやすく、シリカゲルで精製後にほんの少しだがエピマーが観測された。Danishefskyは確かエピマーの方を合成して、これも天然物だ、と言っていた記憶があるが面倒なので文献はチェックしない。残るはt-ブチルエステルをカルボン酸に変換するだけで、ギ酸中室温放置で定量的に(-)-CP-263,114が得られた。