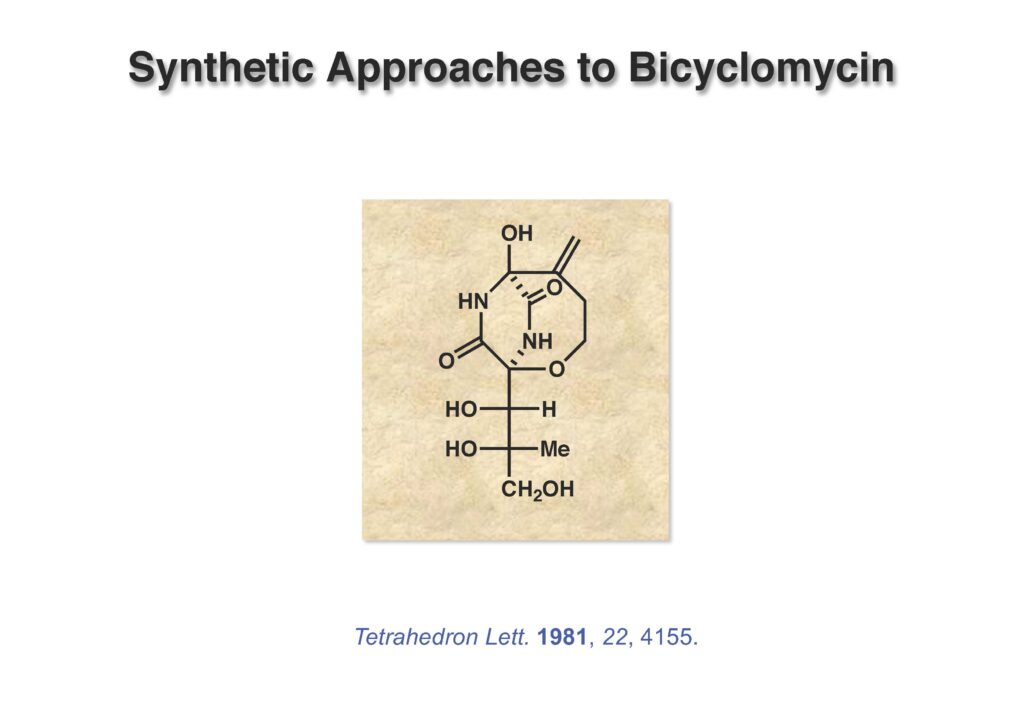
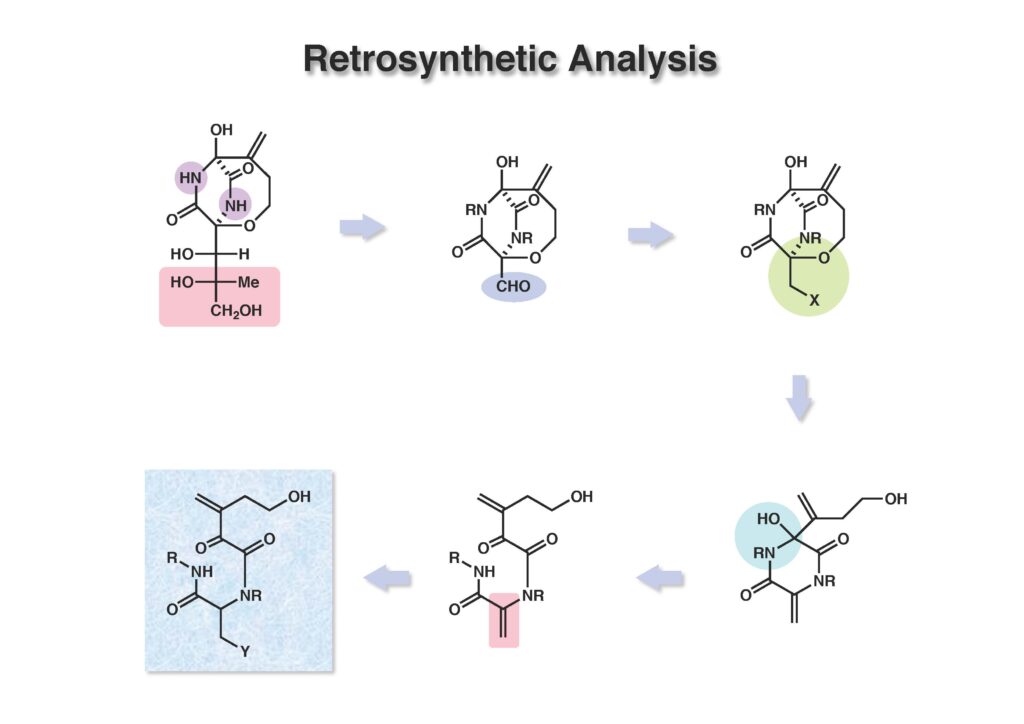
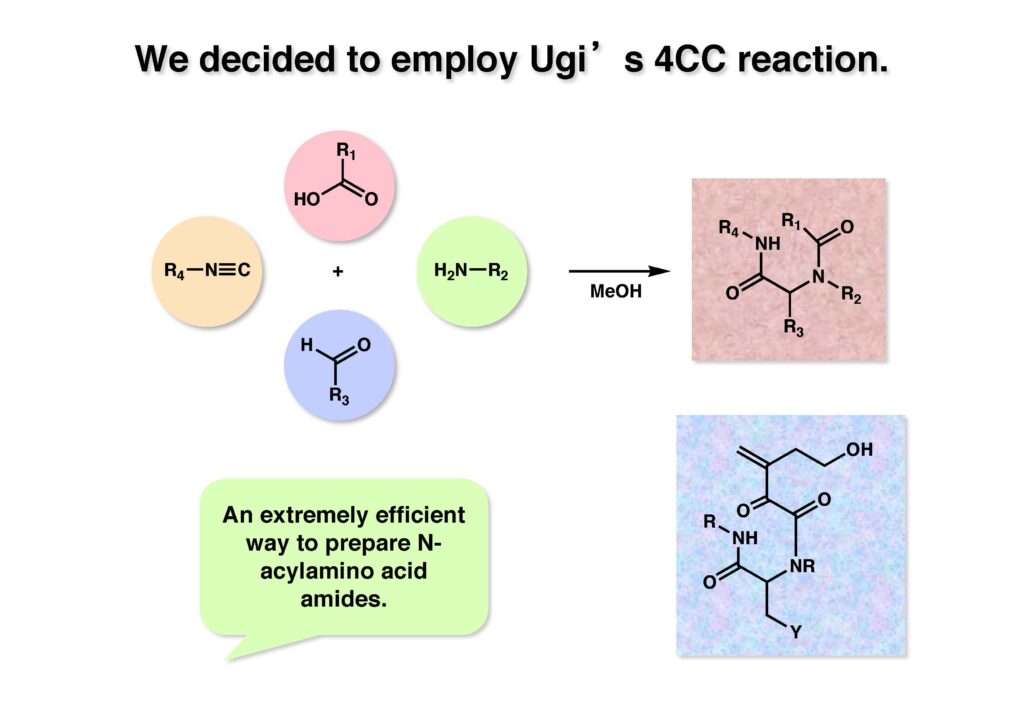
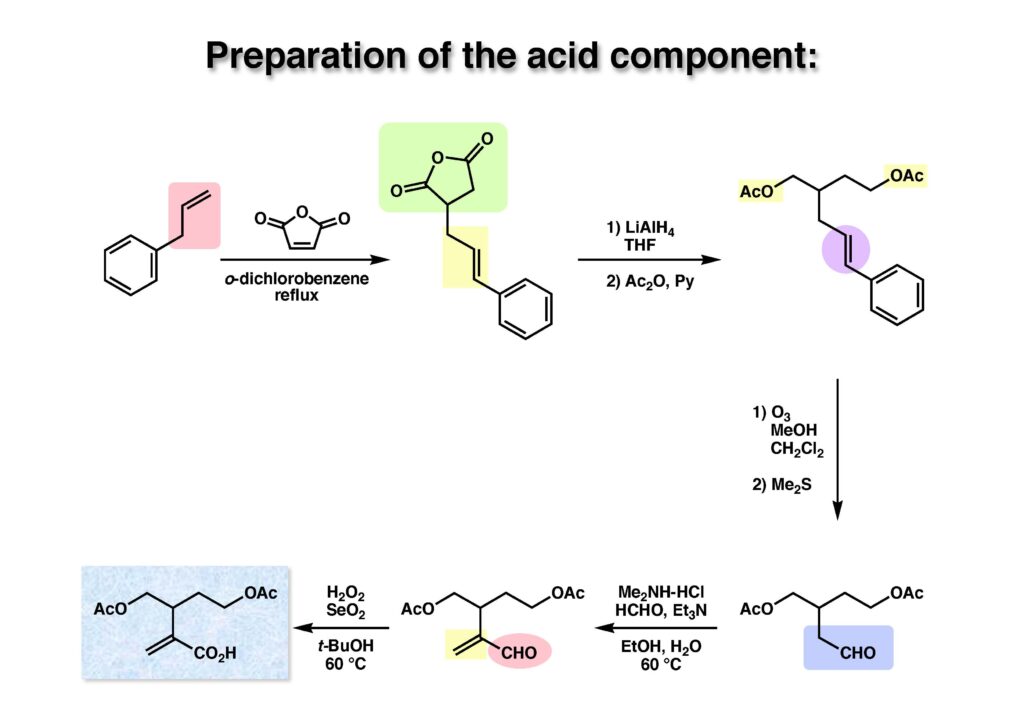
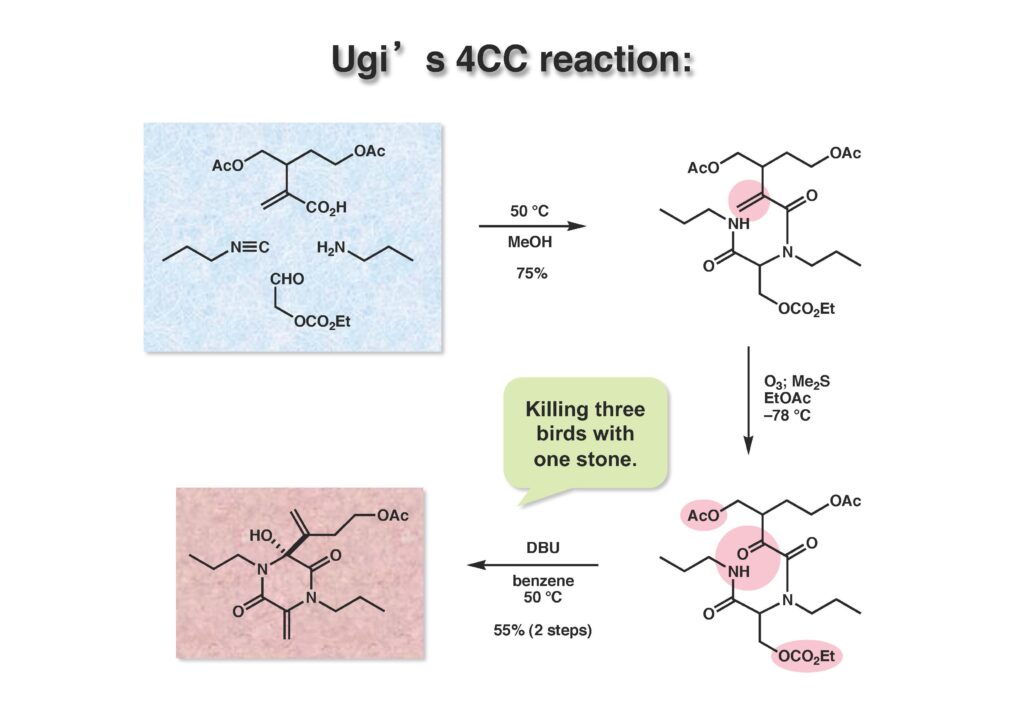
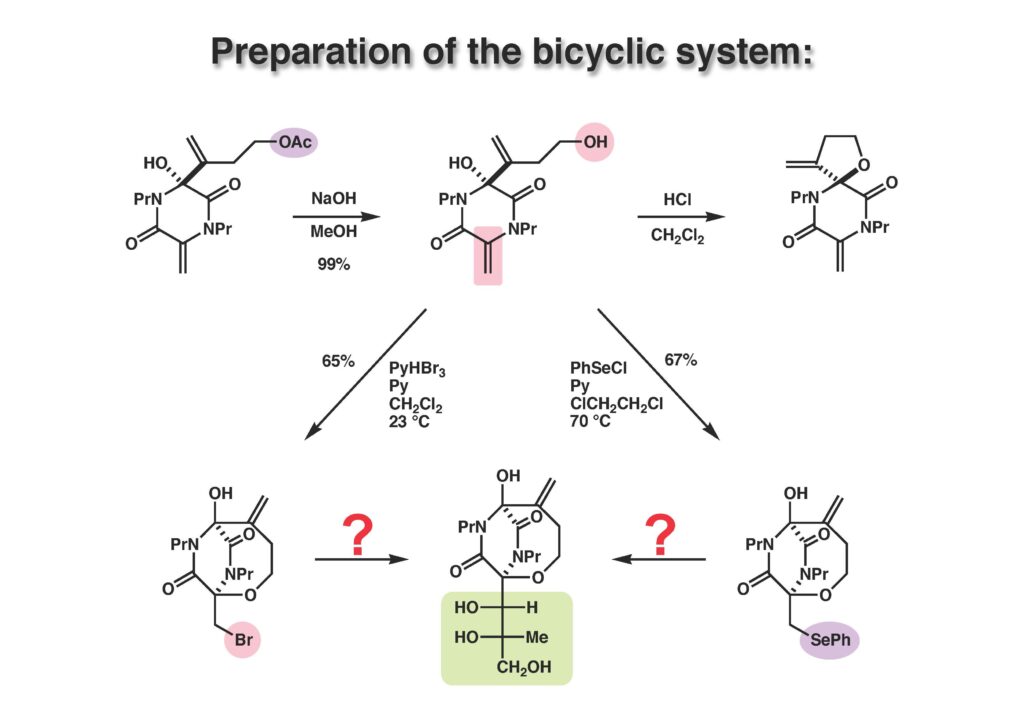
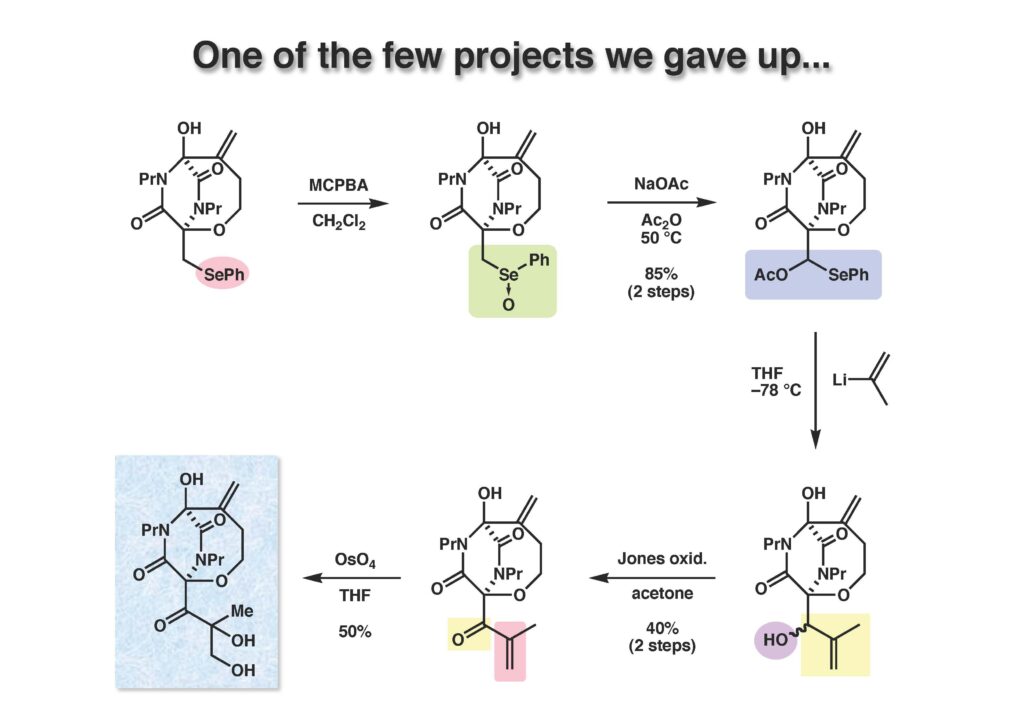
Bicyclomycinは藤沢薬品(現アステラス製薬)で単離構造決定された特異な抗生物質で、グラム陰性菌にも効果があって上市が期待されていたが、残念ながら薬にはならなかった。名前の通り非常にユニークな構造を有しており何とか全合成しようと始めたのだが、学生がもう博士号を取るには十分仕事をやったから、と私から言わせればギブアップ宣言をしたので、その後戸棚に放置してホコリが被った状態が続き、本人もだんだんやる気が無くなって全合成を諦めた情けないプロジェクトである。ただ、考え方としてはUgi反応を利用した面白いルートで窒素の保護基さえ工夫できたら全合成できたはずだと思っている。Ivar Ugi先生にドイツでお会いした時に、この仕事を高く評価されていたのが思い出の一つである。
“Synthetic Approach to Bicyclomycin: Synthesis of the Bicyclic System of Bicyclomycin,” T. Fukuyama, B. D. Robins, and R. A. Sachleben, Tetrahedron Lett., 22, 4155 (1981).