1978年夏にRice大学に赴任して最初に全合成を試みたプロジェクトである。比較的低毒性でMerck社が抗がん剤として開発しようとしたが生産菌が変異してしまい593Aを作らなくなってしまったので開発が頓挫した。私が全合成を開始した時点では少なくとも5グループが全合成に着手していたが 現在までに全合成に成功しているのは私のグループだけである。簡単そうな構造に見えるがなかなか手強い化合物であった。ピヨピヨの大学院1年生と学部生一人ずつのチームで実験はこうやるものだと教えながら1年ちょっとかけて全合成が完成した、まあ、私が一人で全合成したようなものである(反応は全て私が見つけて、学生に後追いさせた)。(笑)
“Total Synthesis of dl-Antibiotic 593A,” T. Fukuyama, R. K. Frank, and C. F. Jewell, Jr., J. Am. Chem. Soc., 102, 2122 (1980).
 Antibiotic 593Aはmitomycin Cと同じくDNAのdouble strandの両方をアルキル化して複製を阻止する作用機構を持っている。アルキル化の機構としてはnitrogen mustardと同じく、アミンの孤立電子対がClを追い出してアジリジン構造となりそれがDNAを攻撃すると考えられている。593Aは水存在下で加熱すると分解してClがOHに変わった化合物となり活性を失うが、その際にdouble inversionが起きるのでOH体の立体化学はCl体と同じである。
Antibiotic 593Aはmitomycin Cと同じくDNAのdouble strandの両方をアルキル化して複製を阻止する作用機構を持っている。アルキル化の機構としてはnitrogen mustardと同じく、アミンの孤立電子対がClを追い出してアジリジン構造となりそれがDNAを攻撃すると考えられている。593Aは水存在下で加熱すると分解してClがOHに変わった化合物となり活性を失うが、その際にdouble inversionが起きるのでOH体の立体化学はCl体と同じである。
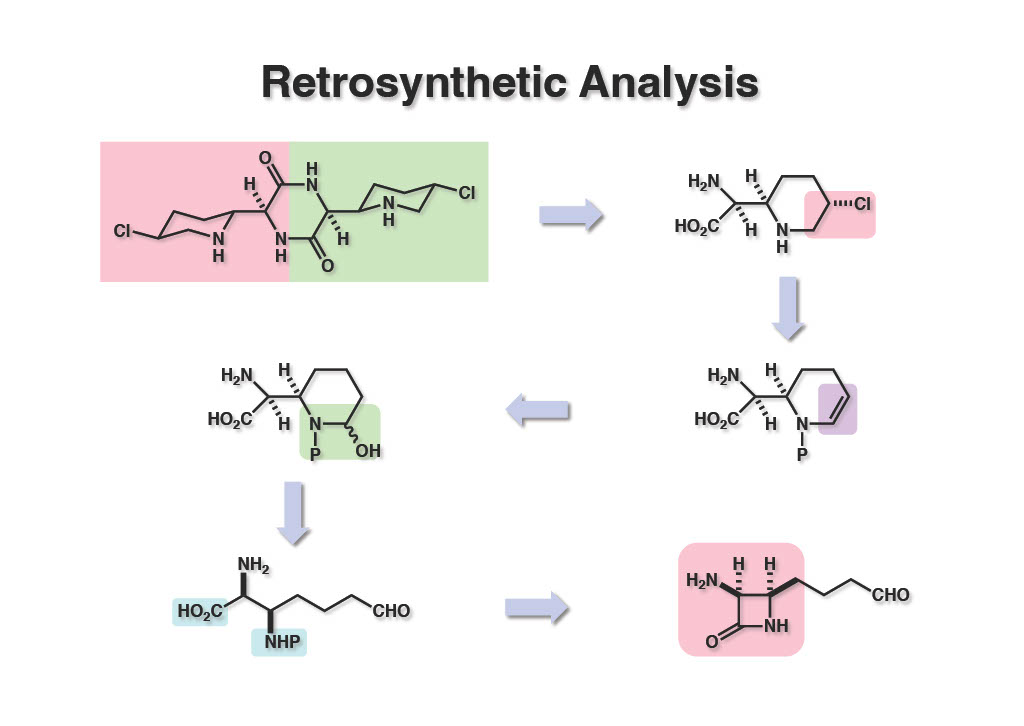 化合物に番号を振っていないので、1列目の左から1番目の化合物には1-1と振っておくのでよろしく。目的化合物 (1-1) はアミノ酸のダイマーだが、このアミノ酸の単体 (1-2) はまだ発見されていない。ピペリジンの3位の塩素はエナミン誘導体 (2-2) への塩素化で導入できると考え、直鎖のα,β-ジアミノ酸 (3-1) の立体化学をいかに制御するかというところで、シス型のβ-ラクタム (3-2) を合成すれば良いという結論に達した。
化合物に番号を振っていないので、1列目の左から1番目の化合物には1-1と振っておくのでよろしく。目的化合物 (1-1) はアミノ酸のダイマーだが、このアミノ酸の単体 (1-2) はまだ発見されていない。ピペリジンの3位の塩素はエナミン誘導体 (2-2) への塩素化で導入できると考え、直鎖のα,β-ジアミノ酸 (3-1) の立体化学をいかに制御するかというところで、シス型のβ-ラクタム (3-2) を合成すれば良いという結論に達した。
 ここで問題となったのは、cis-β-ラクタムを如何にして合成するか、ということと、β-ラクタムのNH体を効率よく合成することであった。Staudingerのβ-ラクタム合成法は主としてaromatic aldehydeとarylamineと種々の酸クロリドを用いており、aliphaticな側鎖を持つ化合物ほ殆ど知られていなかった。β-ラクタム合成後にNH体に変換するのも電子豊富な芳香環をオゾン分解するくらいしか見つからなかった。折よく、PhiladelphiaにあったSKF社(Smith Kline & French)という製薬会社(現在はGlaxoと合併してGSK)から2,4-dimethoxybenzylamineを使えば酸化的に除去してNH体が得られるという報告がJACSに報告されていた。始めはCO2Me基 (2-3) から側鎖を伸ばそうとしたがうまく行かなかったので直接C4個の側鎖を持つβ-ラクタムを合成することにした。
ここで問題となったのは、cis-β-ラクタムを如何にして合成するか、ということと、β-ラクタムのNH体を効率よく合成することであった。Staudingerのβ-ラクタム合成法は主としてaromatic aldehydeとarylamineと種々の酸クロリドを用いており、aliphaticな側鎖を持つ化合物ほ殆ど知られていなかった。β-ラクタム合成後にNH体に変換するのも電子豊富な芳香環をオゾン分解するくらいしか見つからなかった。折よく、PhiladelphiaにあったSKF社(Smith Kline & French)という製薬会社(現在はGlaxoと合併してGSK)から2,4-dimethoxybenzylamineを使えば酸化的に除去してNH体が得られるという報告がJACSに報告されていた。始めはCO2Me基 (2-3) から側鎖を伸ばそうとしたがうまく行かなかったので直接C4個の側鎖を持つβ-ラクタムを合成することにした。
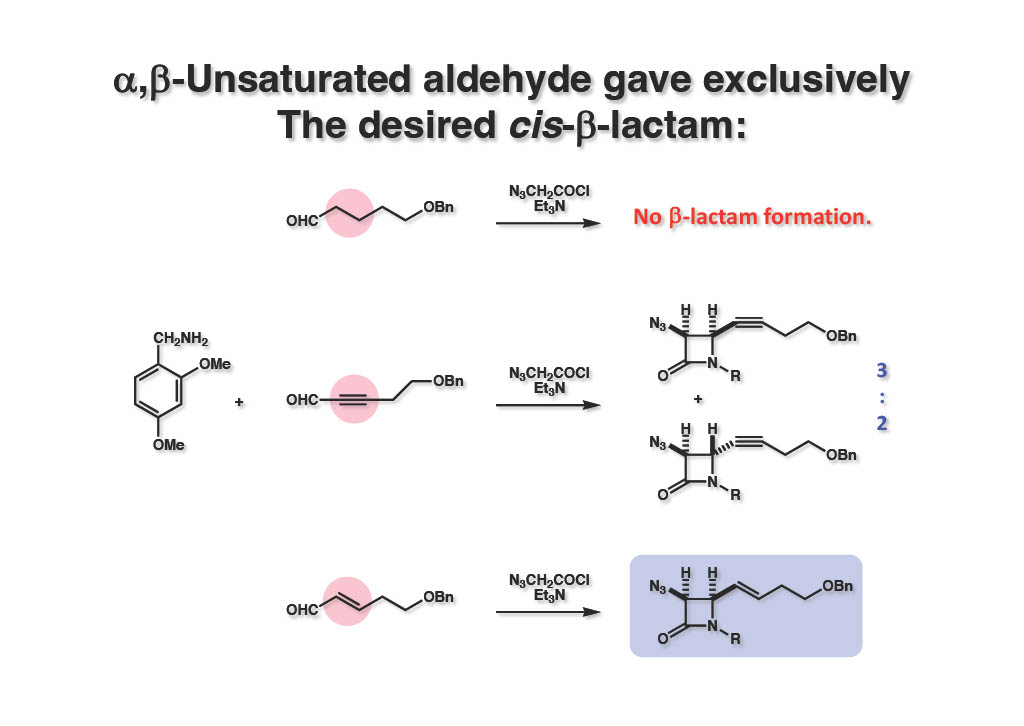 まず、飽和アルデヒド (1-1) を基質にStaudinger反応を試みたが、β-ラクタムは全く得られずにアミンがアミドに変換されただけであった。(後年、α位に酸素原子など電子吸引性の官能基が付いたアルデヒドではβ-ラクタムが合成できるという報告が見られた。)次に合成の順番から(propargyl alcoholを原料にしていたので)三重結合を持つアルデヒド (2-2) を用いたところcis:trans β-ラクタム (2-3) が3:2の比率で得られた。これでは話にならないので、α,β-不飽和アルデヒド(3-1) を使ったところ、首尾よくcis-体 (3-2) のみが得られてきた。この実験結果はStaudinger反応が4電子環状反応を経由するという仮説を考慮に入れると理解できると思う。学生諸君も反応機構を考えてみることを勧めておきたい。
まず、飽和アルデヒド (1-1) を基質にStaudinger反応を試みたが、β-ラクタムは全く得られずにアミンがアミドに変換されただけであった。(後年、α位に酸素原子など電子吸引性の官能基が付いたアルデヒドではβ-ラクタムが合成できるという報告が見られた。)次に合成の順番から(propargyl alcoholを原料にしていたので)三重結合を持つアルデヒド (2-2) を用いたところcis:trans β-ラクタム (2-3) が3:2の比率で得られた。これでは話にならないので、α,β-不飽和アルデヒド(3-1) を使ったところ、首尾よくcis-体 (3-2) のみが得られてきた。この実験結果はStaudinger反応が4電子環状反応を経由するという仮説を考慮に入れると理解できると思う。学生諸君も反応機構を考えてみることを勧めておきたい。
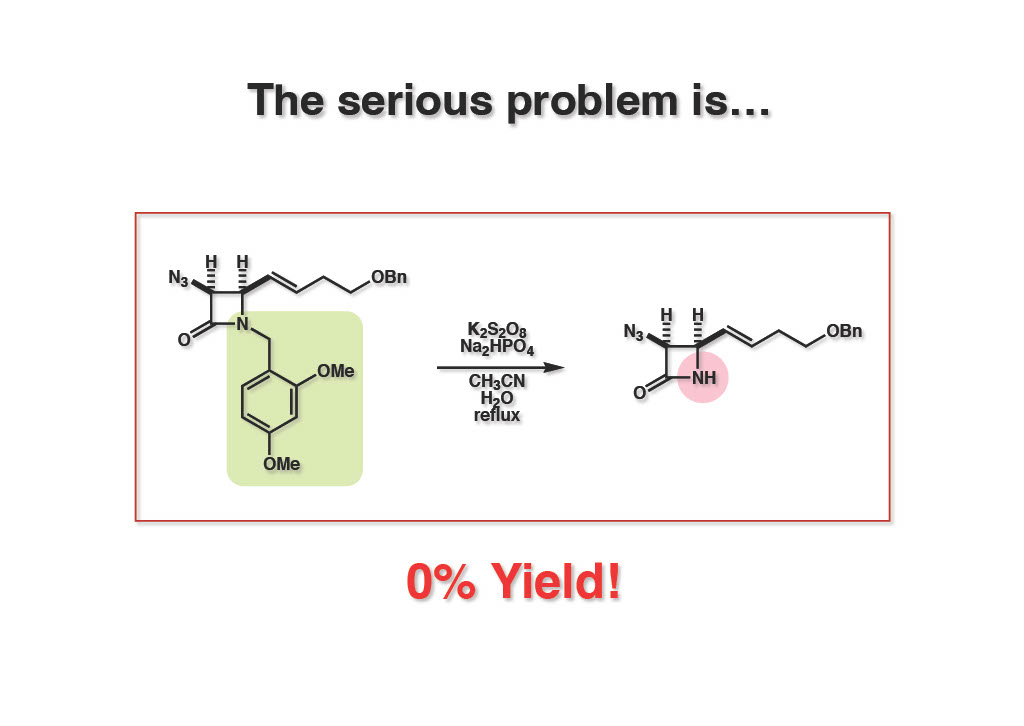 残念ながら2,4-dimethoxybenzyl基は文献条件下では全く望みのNH体 (1-2) を得ることができなかった。二重結合を水添後するとアジド部分はアミンになるのでベンゾイル化して、飽和体を同条件下に付したところ、低収率ながらNH体が彫られてきたが、実用に供するまでには至らなかった。
残念ながら2,4-dimethoxybenzyl基は文献条件下では全く望みのNH体 (1-2) を得ることができなかった。二重結合を水添後するとアジド部分はアミンになるのでベンゾイル化して、飽和体を同条件下に付したところ、低収率ながらNH体が彫られてきたが、実用に供するまでには至らなかった。
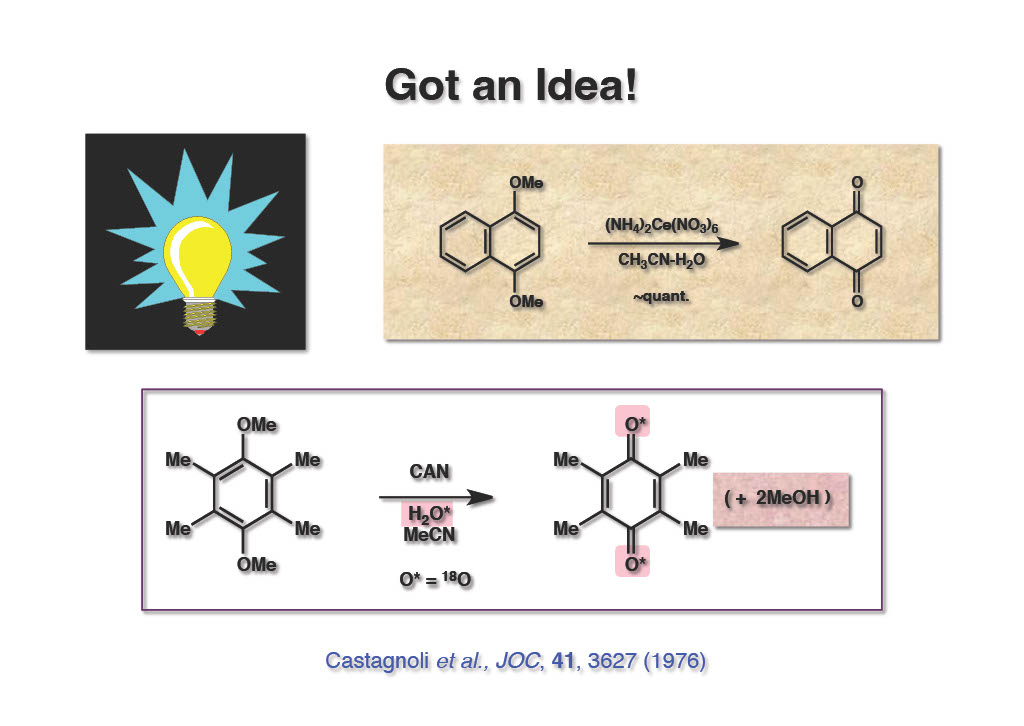 その後、H2NCH2CH2SePh、allylamine(Tetrahedron Lett., 25, 4709 (1984))、H2NCH2CH(OEt)2などを用いることで数段階でNH体のβ-ラクタムを合成することに成功したが、非効率的でダサい方法だったので実用に供する気持ちは薄かった。ある日、JOCを何となくペラペラとめくっていたところ、CastagnoliのCANによるキノン合成法が私の目に留まった。待てよ、これはアミンやアミドそしてアルコールの保護基になるかもと直感したのである。ラベル化実験で水の酸素がキノンに取り込まれることが示され、メタノールの記述は無かったように思えるがそれは自明のことであるので早速この反応を試してみることにした。
その後、H2NCH2CH2SePh、allylamine(Tetrahedron Lett., 25, 4709 (1984))、H2NCH2CH(OEt)2などを用いることで数段階でNH体のβ-ラクタムを合成することに成功したが、非効率的でダサい方法だったので実用に供する気持ちは薄かった。ある日、JOCを何となくペラペラとめくっていたところ、CastagnoliのCANによるキノン合成法が私の目に留まった。待てよ、これはアミンやアミドそしてアルコールの保護基になるかもと直感したのである。ラベル化実験で水の酸素がキノンに取り込まれることが示され、メタノールの記述は無かったように思えるがそれは自明のことであるので早速この反応を試してみることにした。
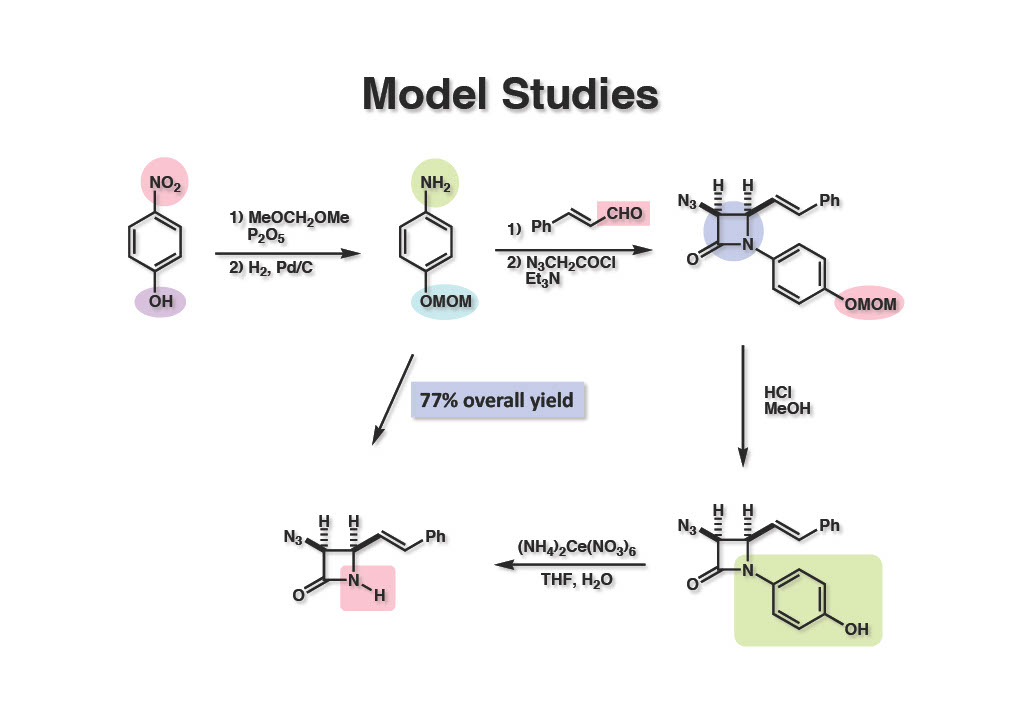 p-Nitrophenol (1-1) をMOM化、水添して得られたアミン (1-2) とcinnamaldehydeを縮合させ、N3CH2COCl(爆発性あり!!合成しようと思わないこと!)とEt3Nでケテンを発生させて反応させると、望むcis体のβ-ラクタム (1-3) のみが得られた。MOM基を除去後にCANで酸化すると高収率でNH体 (2-1) を合成することができた。Staudinger反応でNH体を合成する実用的な保護基の先駆的な方法であった。
p-Nitrophenol (1-1) をMOM化、水添して得られたアミン (1-2) とcinnamaldehydeを縮合させ、N3CH2COCl(爆発性あり!!合成しようと思わないこと!)とEt3Nでケテンを発生させて反応させると、望むcis体のβ-ラクタム (1-3) のみが得られた。MOM基を除去後にCANで酸化すると高収率でNH体 (2-1) を合成することができた。Staudinger反応でNH体を合成する実用的な保護基の先駆的な方法であった。
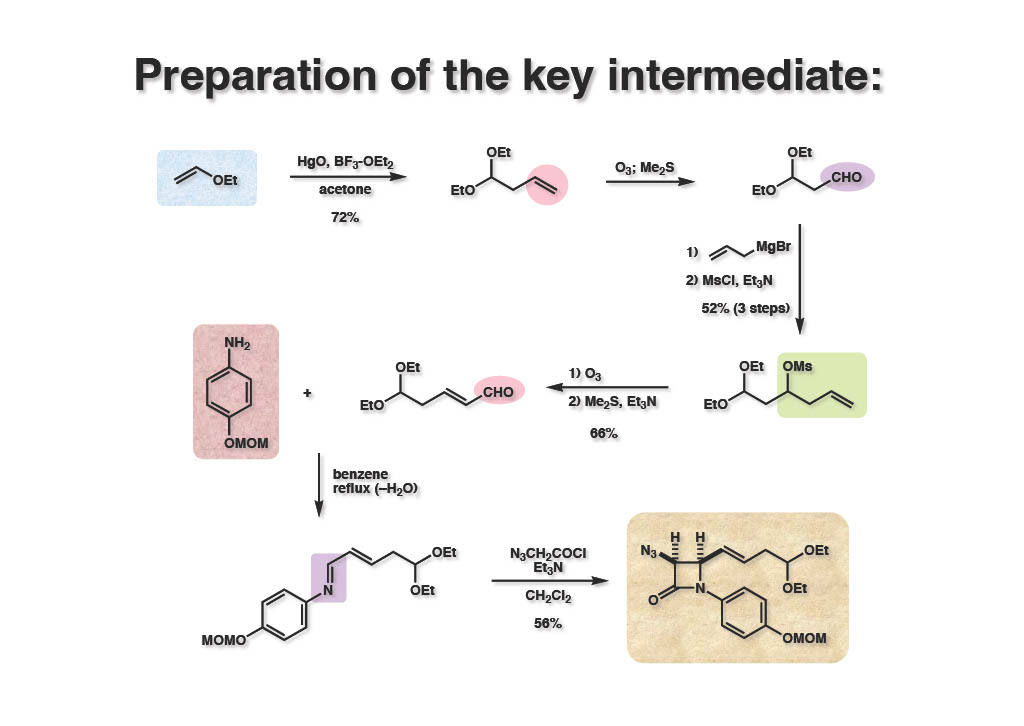 全合成を開始するにあたり、まずマロンジアルデヒドの一方のアルデヒドをアセタールで保護した化合物 (2-2) を合成する必要があった。(MeO)2CHCH2CH(OMe)2は非常に安価な化合物であるが、片方のアルデヒドだけ加水分解するのは困難であり、実用的ではなかった。そこで文献既知の3-butenalのジエチルアセタール (1-2) をエチルビニルエーテル (1-1) の2量化で合成した。酸化水銀は2種類の結晶体が市販されているが、どちらか一方を使わないと2量化は成功しなかった(どちらがどちらかは忘却の彼方へ)。これをPh3P=CHCHOでWittig反応に付せば望む化合物 (2-2) が得られるのだが、 ClCH2CHOが水溶液として市販されていたためWittig試薬を作るのが面倒で、多段階ながらも大量合成できる方法を採用した。すなわち、allylmagnesium bromideの付加、生成したアルコールのメシル化、次いでオゾン分解後にMe2SとEt3Nを同時に加えて還元、脱離を行うことにより望むアルデヒド (2-2) を得た。これをStaudinger反応に用いて重要中間体であるcis-β-ラクタムに (3-2) 導いた。
全合成を開始するにあたり、まずマロンジアルデヒドの一方のアルデヒドをアセタールで保護した化合物 (2-2) を合成する必要があった。(MeO)2CHCH2CH(OMe)2は非常に安価な化合物であるが、片方のアルデヒドだけ加水分解するのは困難であり、実用的ではなかった。そこで文献既知の3-butenalのジエチルアセタール (1-2) をエチルビニルエーテル (1-1) の2量化で合成した。酸化水銀は2種類の結晶体が市販されているが、どちらか一方を使わないと2量化は成功しなかった(どちらがどちらかは忘却の彼方へ)。これをPh3P=CHCHOでWittig反応に付せば望む化合物 (2-2) が得られるのだが、 ClCH2CHOが水溶液として市販されていたためWittig試薬を作るのが面倒で、多段階ながらも大量合成できる方法を採用した。すなわち、allylmagnesium bromideの付加、生成したアルコールのメシル化、次いでオゾン分解後にMe2SとEt3Nを同時に加えて還元、脱離を行うことにより望むアルデヒド (2-2) を得た。これをStaudinger反応に用いて重要中間体であるcis-β-ラクタムに (3-2) 導いた。
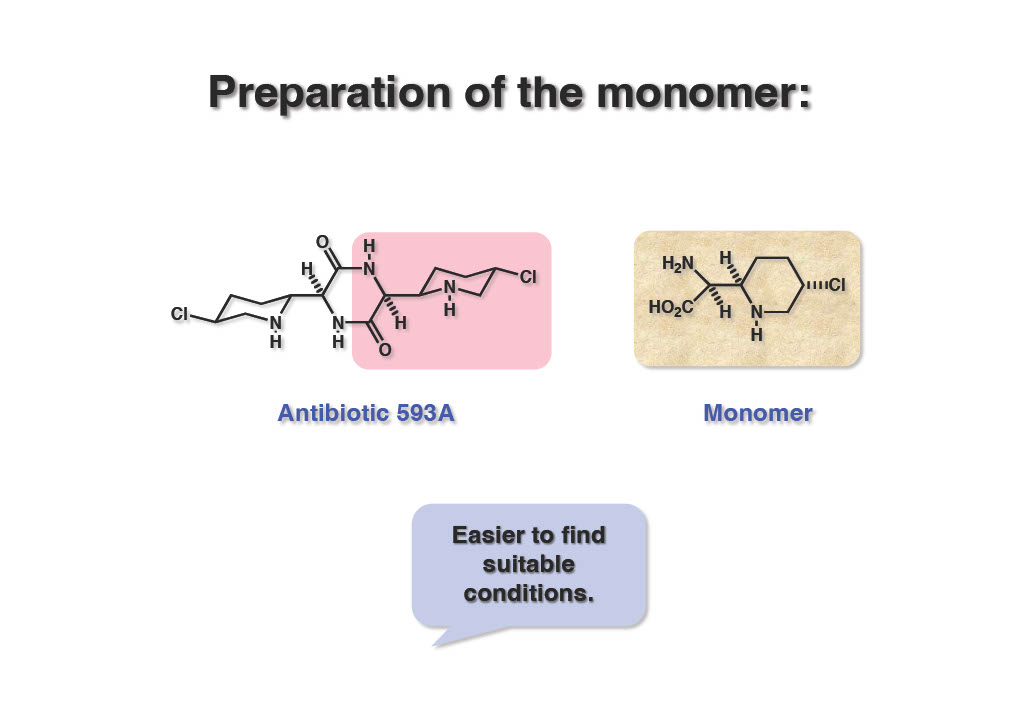 当然のことながら、ダイマーを使って条件検討すれば50%の反応も25%になってしまうので、モノマーを使ってルートの探索を行うことにした。
当然のことながら、ダイマーを使って条件検討すれば50%の反応も25%になってしまうので、モノマーを使ってルートの探索を行うことにした。
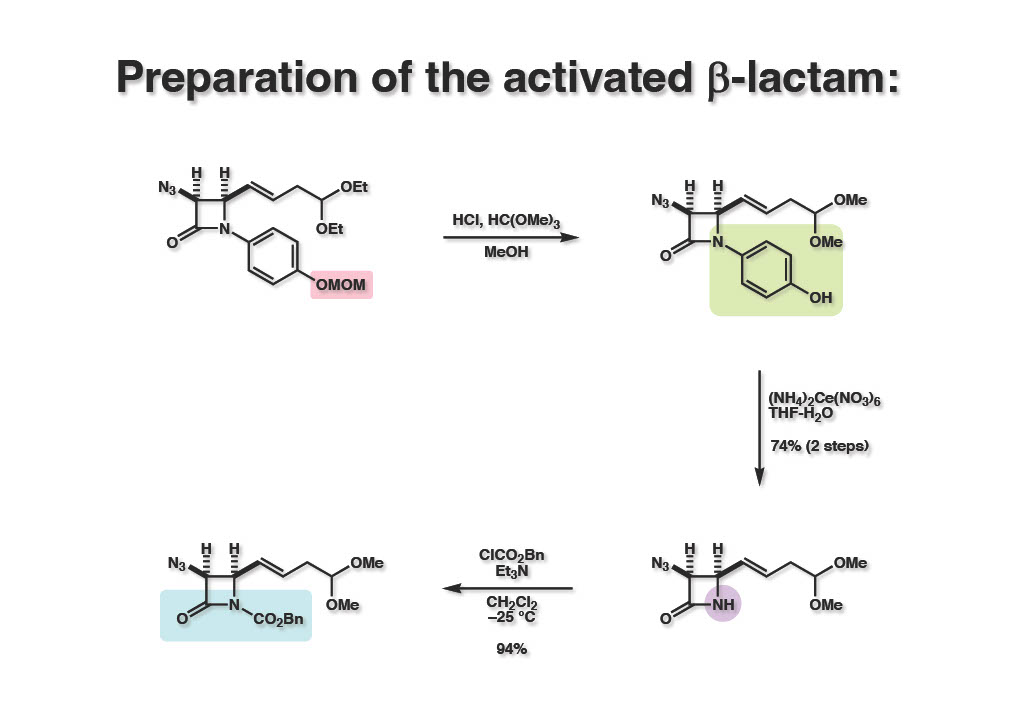 β-ラクタムは加水分解されやすいと言われているが、実際にはそう簡単に塩基性条件で加水分解はされない。そこでNH体 (2-2) を低温下でCbzClと反応させてイミド (2-1) へと変換した。おそらくhybridizationに起因すると思うがβ-ラクタムのアミドは結構反応性が高く、アシル化やシリル化は普通のアミドに比べて容易に進行する。
β-ラクタムは加水分解されやすいと言われているが、実際にはそう簡単に塩基性条件で加水分解はされない。そこでNH体 (2-2) を低温下でCbzClと反応させてイミド (2-1) へと変換した。おそらくhybridizationに起因すると思うがβ-ラクタムのアミドは結構反応性が高く、アシル化やシリル化は普通のアミドに比べて容易に進行する。
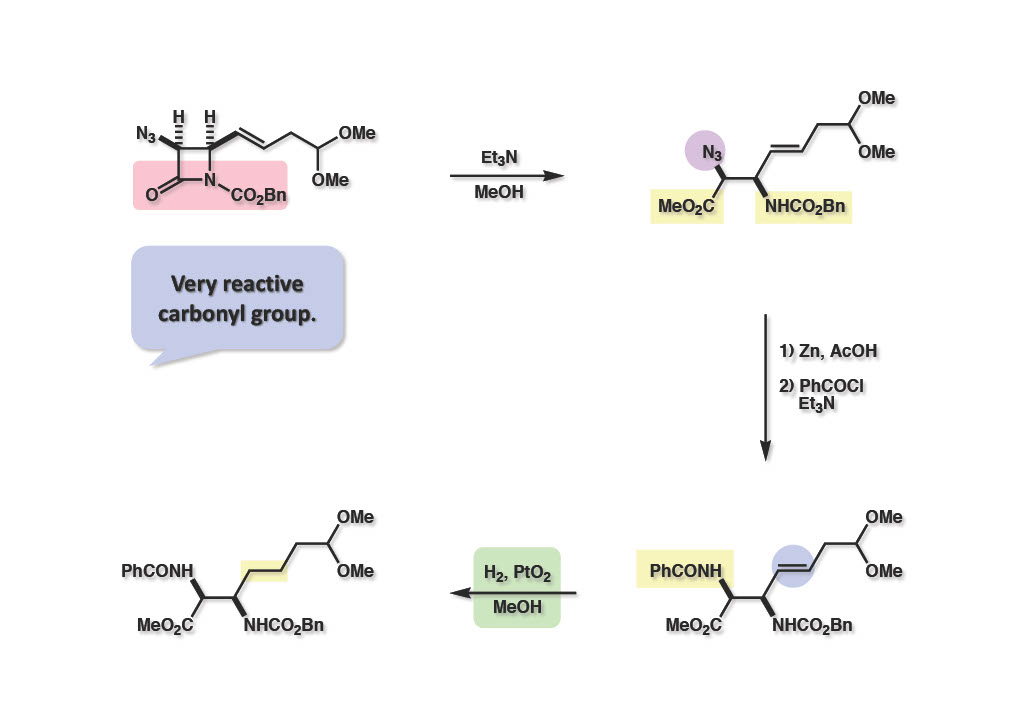 トリエチルアミンを触媒とした加メタノール分解は容易に進行し、メチルエステル (2-2) が得られた。次にアジドを亜鉛還元して生成したアミンをベンゾイル化し、二重結合を水添して飽和体 (2-1) を得た。パラジウム触媒を使うとCbz基が外れてしまうので、hydrogenolysisが遅いAdams触媒を用いたこともコメントしておく。
トリエチルアミンを触媒とした加メタノール分解は容易に進行し、メチルエステル (2-2) が得られた。次にアジドを亜鉛還元して生成したアミンをベンゾイル化し、二重結合を水添して飽和体 (2-1) を得た。パラジウム触媒を使うとCbz基が外れてしまうので、hydrogenolysisが遅いAdams触媒を用いたこともコメントしておく。
 ウレタンとアセタールの環化脱メタノール反応は、生成物が加水分解やカチオン生成による副反応を受けやすいために弱酸性条件で行う必要があった。ここで思い出したのは、シクロヘキサノンのジエチルアセタールを脱エタノール化でエノールエーテルを合成する条件に、p-TsOHとキノリンを用いて、確か50度Cくらいに加温して少し減圧してエタノールを除去するという報告があったことである。p-TsOHの代わりにCSAを用いて反応を行ったところ非常に綺麗にエナミド体 (1-2) (実際はウレタンであるが語呂が悪いので)が得られた。Strong acid-weak base combinationであるが、同様の発想は宮下先生がGrieco研で発見したPPTSにも見られる。TFが岸研で酸に弱いマイトマイシンCの全合成に従事していた時も、PhNMe2-HClO4を用いてβ-(aziridyl)propionaldehydeのretro-Mannich反応を行ったことがある。
ウレタンとアセタールの環化脱メタノール反応は、生成物が加水分解やカチオン生成による副反応を受けやすいために弱酸性条件で行う必要があった。ここで思い出したのは、シクロヘキサノンのジエチルアセタールを脱エタノール化でエノールエーテルを合成する条件に、p-TsOHとキノリンを用いて、確か50度Cくらいに加温して少し減圧してエタノールを除去するという報告があったことである。p-TsOHの代わりにCSAを用いて反応を行ったところ非常に綺麗にエナミド体 (1-2) (実際はウレタンであるが語呂が悪いので)が得られた。Strong acid-weak base combinationであるが、同様の発想は宮下先生がGrieco研で発見したPPTSにも見られる。TFが岸研で酸に弱いマイトマイシンCの全合成に従事していた時も、PhNMe2-HClO4を用いてβ-(aziridyl)propionaldehydeのretro-Mannich反応を行ったことがある。
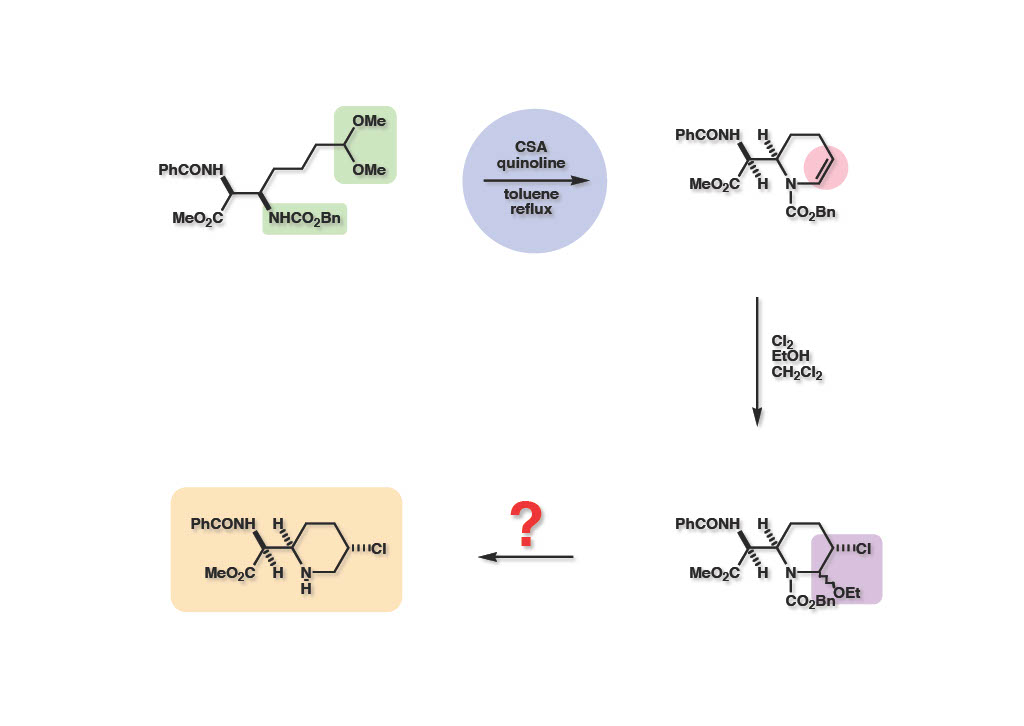
電子豊富なエナミド (1-2) は予想通りエタノール中で塩素を通じることによりジアステレオマー混合物 (2-2) を与えた。次の課題はethoxy基を還元的に除去することであった。
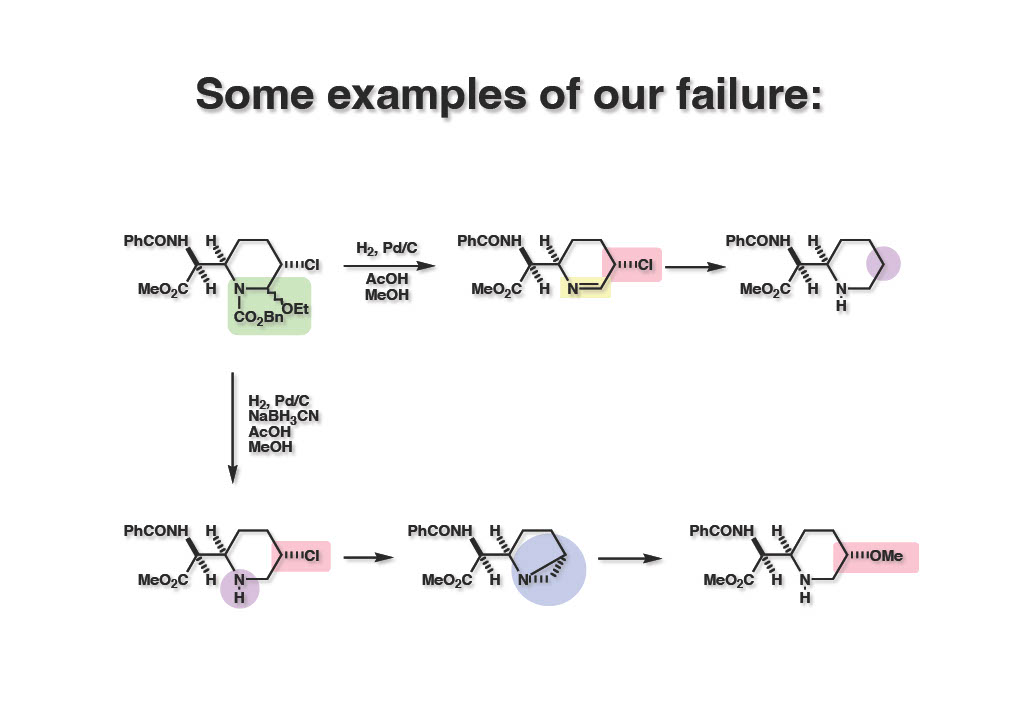 当初は簡単と思われていた脱エトキシ基はそれほど容易ではなかった。Cbz基 (1-1) の加水素分解で得られるイミン (1-2) を同条件下で還元しようとしたらα位のクロル基が先に加水素分解を受けてクロル基の無い生成物 (1-3) が得られてきた。そこでイミン (1-2) をNaBH3CNで還元しようとしたが、おそらくCNイオンが触媒毒となったのか、Cbz基の加水素分解が異常に遅くなり、生成したアミンがクロル基を追い出してアジリジンとなり、溶媒のメタノールが付加したメトキシ体 (2-3) が得られるという間抜けな結果となった。
当初は簡単と思われていた脱エトキシ基はそれほど容易ではなかった。Cbz基 (1-1) の加水素分解で得られるイミン (1-2) を同条件下で還元しようとしたらα位のクロル基が先に加水素分解を受けてクロル基の無い生成物 (1-3) が得られてきた。そこでイミン (1-2) をNaBH3CNで還元しようとしたが、おそらくCNイオンが触媒毒となったのか、Cbz基の加水素分解が異常に遅くなり、生成したアミンがクロル基を追い出してアジリジンとなり、溶媒のメタノールが付加したメトキシ体 (2-3) が得られるという間抜けな結果となった。
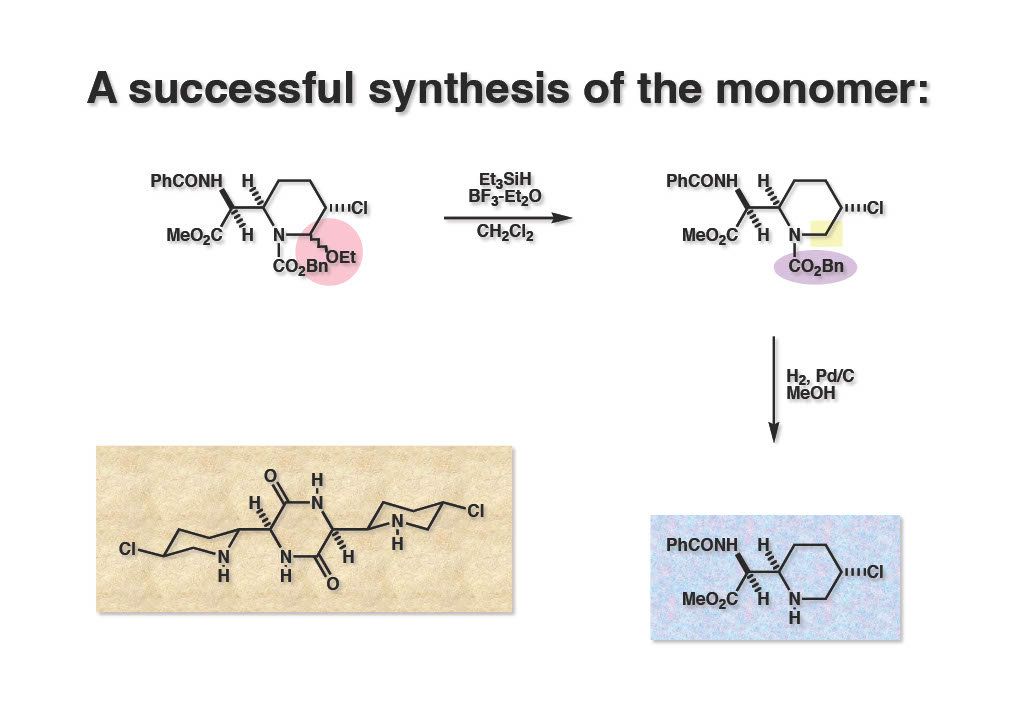 ということで、Cbz基を除去する前にEtO基と還元的に除去しなければならず、酸性条件下でカチオンを還元することができるEt3SiHを用いることにした。メチレンクロライド中、 (1-1) にEt3SiH存在下にBF3・Et2Oを作用させると高収率で脱EtO基が進行して (1-2) が得られた。90MHzのNMRではジアステレオマーの立体化学の解析は困難だったが、脱EtO体は単一物でClが置換されているCHシグナルが18Hzくらいに広がっていたのでC-Cl基は望むequatorial配置であることが判明した。 (1-2) のCbz基を加水素分解してモノマー体 (2-2) が得られた。これでモノマーの合成ルートは確立したので次はダイマーの合成に取り掛かった。
ということで、Cbz基を除去する前にEtO基と還元的に除去しなければならず、酸性条件下でカチオンを還元することができるEt3SiHを用いることにした。メチレンクロライド中、 (1-1) にEt3SiH存在下にBF3・Et2Oを作用させると高収率で脱EtO基が進行して (1-2) が得られた。90MHzのNMRではジアステレオマーの立体化学の解析は困難だったが、脱EtO体は単一物でClが置換されているCHシグナルが18Hzくらいに広がっていたのでC-Cl基は望むequatorial配置であることが判明した。 (1-2) のCbz基を加水素分解してモノマー体 (2-2) が得られた。これでモノマーの合成ルートは確立したので次はダイマーの合成に取り掛かった。
 アミノ酸のダイマー化でジケトピペラジンを合成するのは容易だと、たかを括っていたが、実際は困難を極めた。まず、アミノ酸エステル (1-1) を高温で加熱したり、2-hydroxypyridine中で加熱したが、全くジケトピペラジン (1-2) は生成しなかった。そこでアミド (2-1) を作って加熱したが、これも環化しなかった。おそらくアミノ酸に加水分解して縮合剤を使えば環化は成功したかもしれないが、別のアイデアが浮かんだのでそちらを試すことにした。
アミノ酸のダイマー化でジケトピペラジンを合成するのは容易だと、たかを括っていたが、実際は困難を極めた。まず、アミノ酸エステル (1-1) を高温で加熱したり、2-hydroxypyridine中で加熱したが、全くジケトピペラジン (1-2) は生成しなかった。そこでアミド (2-1) を作って加熱したが、これも環化しなかった。おそらくアミノ酸に加水分解して縮合剤を使えば環化は成功したかもしれないが、別のアイデアが浮かんだのでそちらを試すことにした。
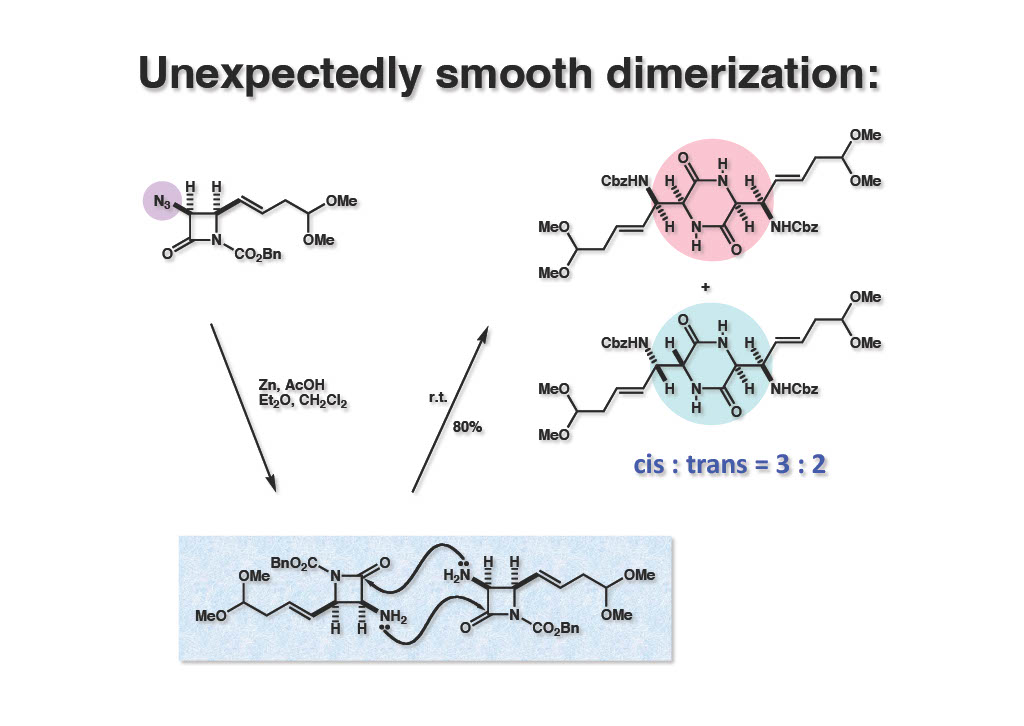 Cbz基で活性化したβ-ラクタム (1-1) のアジド基を亜鉛還元し、後処理後に溶媒をほぼ除いた状態で室温放置したところ、cis体 (1-2) とtrans体 (2-1) のジケトピペラジンが3:2の比率で収率よく得られた。Trans体の生成は出発物がラセミ体なので当然の結果である。もちろん、モノマーが光学活性体であればcis体しか得られないはずである。
Cbz基で活性化したβ-ラクタム (1-1) のアジド基を亜鉛還元し、後処理後に溶媒をほぼ除いた状態で室温放置したところ、cis体 (1-2) とtrans体 (2-1) のジケトピペラジンが3:2の比率で収率よく得られた。Trans体の生成は出発物がラセミ体なので当然の結果である。もちろん、モノマーが光学活性体であればcis体しか得られないはずである。
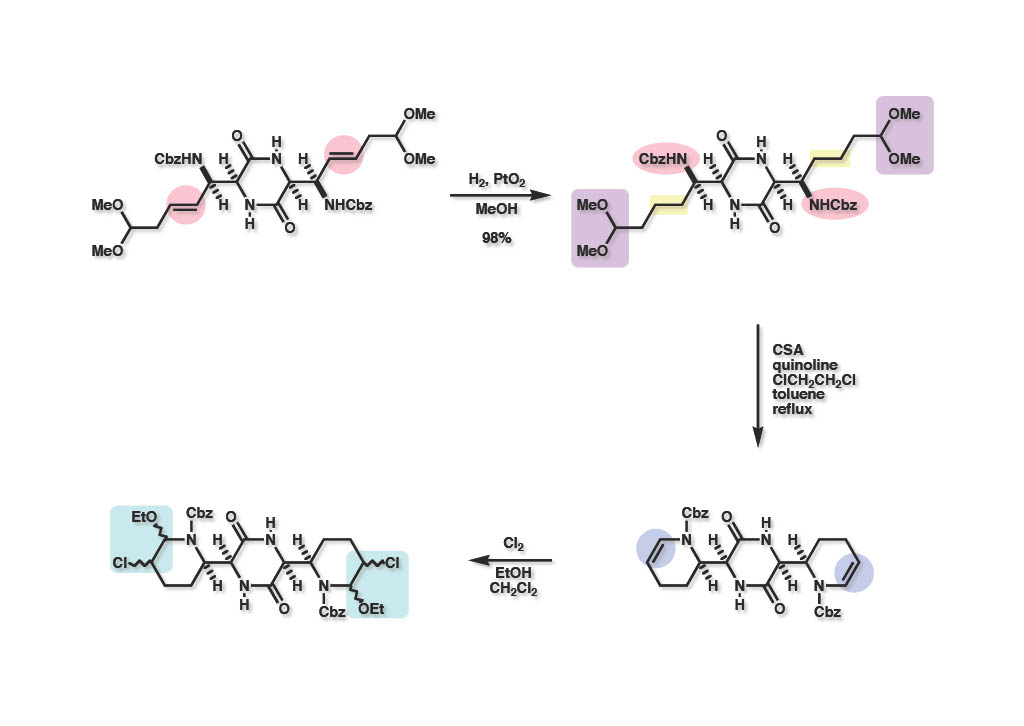 Cis体 (1-1) をAdams触媒を用いて接触還元し、CSA-quinoline存在下で加熱するとエナミド体 (2-2) が高収率で得られた。ここでEtOH存在下に塩素ガスを通じたところNMR的には複雑な混合物 (2-1) が得られてきた。
Cis体 (1-1) をAdams触媒を用いて接触還元し、CSA-quinoline存在下で加熱するとエナミド体 (2-2) が高収率で得られた。ここでEtOH存在下に塩素ガスを通じたところNMR的には複雑な混合物 (2-1) が得られてきた。
 この混合物 (1-1) をモノマーの時と同様にEt3SiHとBF3・Et2Oで処理したところ単一の化合物にはならず、Cbz基を外すまでもなく他の方法を試してみることにした。即ち、上記混合物をC H2Cl2中でBCl3処理をするとCbz基は除去され、EtO基はCl基に置換されるはずである。試薬を減圧で除去してメタノールを加えると、おそらくbis-iminium塩 (2-1) が生成するはずであり、これを還元するためにNaBH3CNと念の為に酢酸も加えたところかなりきれいな生成物が得られた。NMRを測定したところ天然物と一致し、結晶としては500 mgほど合成することができた。望む化合物が主生成物となったのは、おそらくiminium塩の段階でenamineとの平衡反応が起き、熱力学的に有利なpseudo-equatorial型配置 (3-1) が優勢となり、それが還元されることにより593Aが主生成物として得られたと思う。
この混合物 (1-1) をモノマーの時と同様にEt3SiHとBF3・Et2Oで処理したところ単一の化合物にはならず、Cbz基を外すまでもなく他の方法を試してみることにした。即ち、上記混合物をC H2Cl2中でBCl3処理をするとCbz基は除去され、EtO基はCl基に置換されるはずである。試薬を減圧で除去してメタノールを加えると、おそらくbis-iminium塩 (2-1) が生成するはずであり、これを還元するためにNaBH3CNと念の為に酢酸も加えたところかなりきれいな生成物が得られた。NMRを測定したところ天然物と一致し、結晶としては500 mgほど合成することができた。望む化合物が主生成物となったのは、おそらくiminium塩の段階でenamineとの平衡反応が起き、熱力学的に有利なpseudo-equatorial型配置 (3-1) が優勢となり、それが還元されることにより593Aが主生成物として得られたと思う。
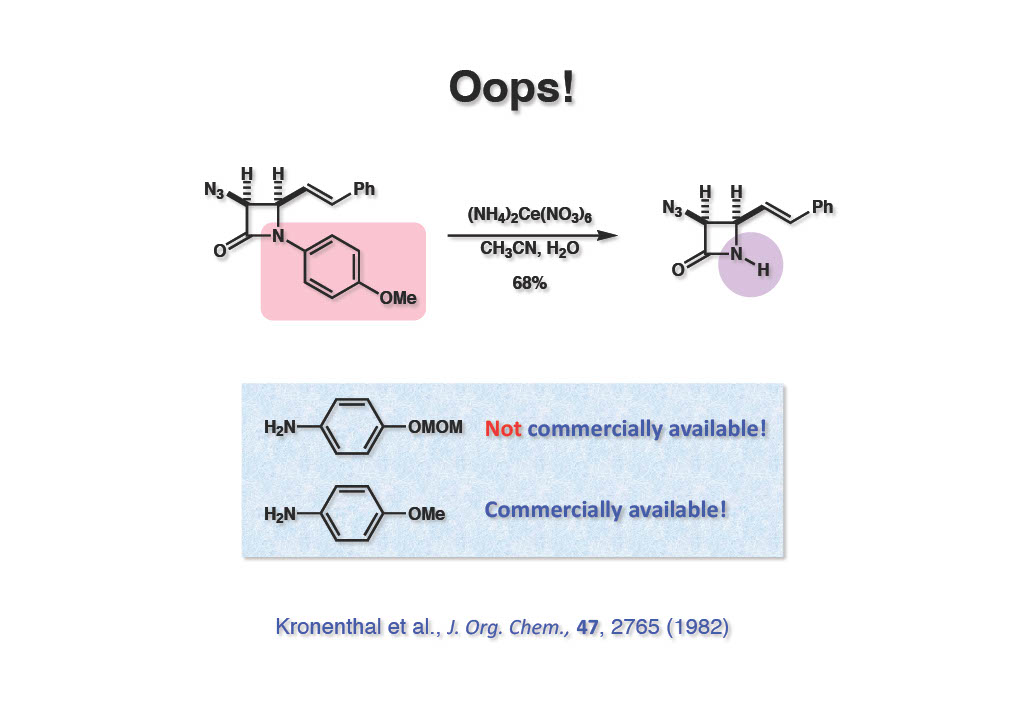 これで一件落着ということで、少人数の研究室としては別のプロジェクトを開始していたが、JOCにSquibb社(現在はBristol Myers Squibb)の Kronenthal(Stork研出身)が、より実用的なβ-ラクタムの保護基としてp-anisidineを用いれば良いということを報告されてしまった。実は上記の反応は正しく私たちが最初に試した反応で、MeOよりもOHの方がCAN酸化の収率が高かったので、593A合成ではMOM基で保護したものを使っていた。その後英国の製薬会社の連中がMOM基を付けたままCANで脱保護した特許報告を見てKronenthalは、それなら市販品のp-anisidineを使えば良い、ということになったのだと思う。今ではこちらが主流で、元祖は忘れ去られているのが残念である。まあ、論文を出さなかったお前が悪いって言われれば、そのとおり!
これで一件落着ということで、少人数の研究室としては別のプロジェクトを開始していたが、JOCにSquibb社(現在はBristol Myers Squibb)の Kronenthal(Stork研出身)が、より実用的なβ-ラクタムの保護基としてp-anisidineを用いれば良いということを報告されてしまった。実は上記の反応は正しく私たちが最初に試した反応で、MeOよりもOHの方がCAN酸化の収率が高かったので、593A合成ではMOM基で保護したものを使っていた。その後英国の製薬会社の連中がMOM基を付けたままCANで脱保護した特許報告を見てKronenthalは、それなら市販品のp-anisidineを使えば良い、ということになったのだと思う。今ではこちらが主流で、元祖は忘れ去られているのが残念である。まあ、論文を出さなかったお前が悪いって言われれば、そのとおり!
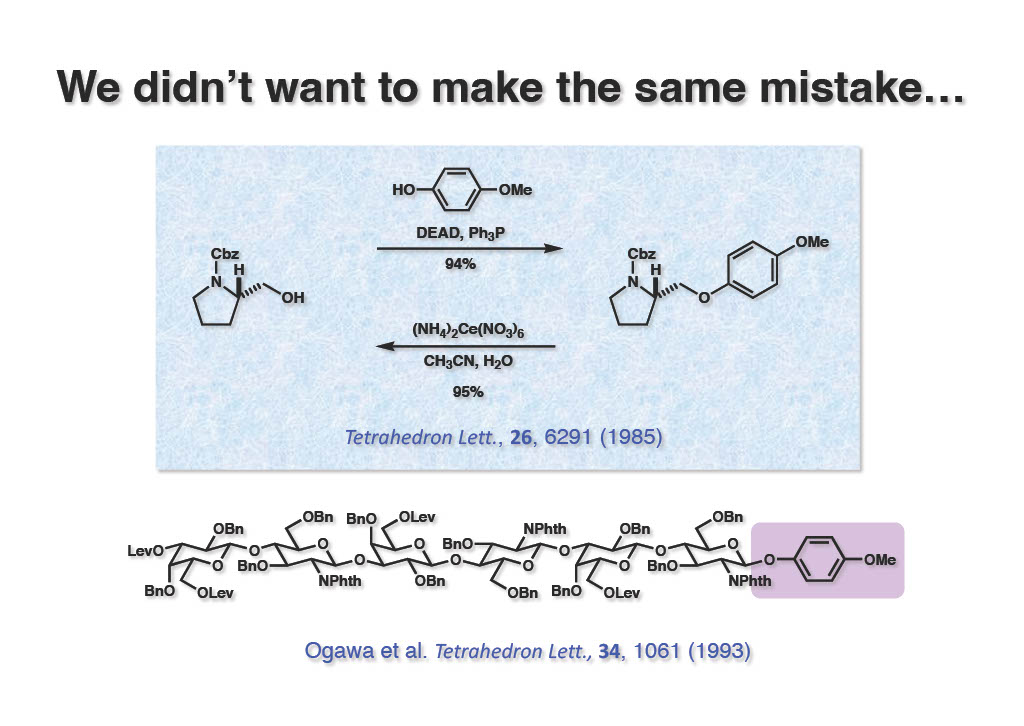 そこで、既に研究室では使っていたp-methoxyphenyl基の論文を急いで出すことにした。保護基としては第一級アルコールにしか使えないが、強酸性、強塩基性、還元、通常の酸化にも耐え、必要な時にはCANであっという間に除去できる、知る人ぞ知る保護基である。全合成の初期段階に導入して、最終段階で除去することができるので、ニッチ的ながらも有用な保護基であると思う。理研の小川智也先生は多糖類の合成に我々の保護基を使って頂いた。このAntibiotic 593Aの全合成はTFが独立して最初に挑んだ仕事で、β-ラクタムを使った最初の全合成であり、β-ラクタムの新規保護基の開発にも成功し、私の研究成果の中ではかなり高いランクに位置している。
そこで、既に研究室では使っていたp-methoxyphenyl基の論文を急いで出すことにした。保護基としては第一級アルコールにしか使えないが、強酸性、強塩基性、還元、通常の酸化にも耐え、必要な時にはCANであっという間に除去できる、知る人ぞ知る保護基である。全合成の初期段階に導入して、最終段階で除去することができるので、ニッチ的ながらも有用な保護基であると思う。理研の小川智也先生は多糖類の合成に我々の保護基を使って頂いた。このAntibiotic 593Aの全合成はTFが独立して最初に挑んだ仕事で、β-ラクタムを使った最初の全合成であり、β-ラクタムの新規保護基の開発にも成功し、私の研究成果の中ではかなり高いランクに位置している。