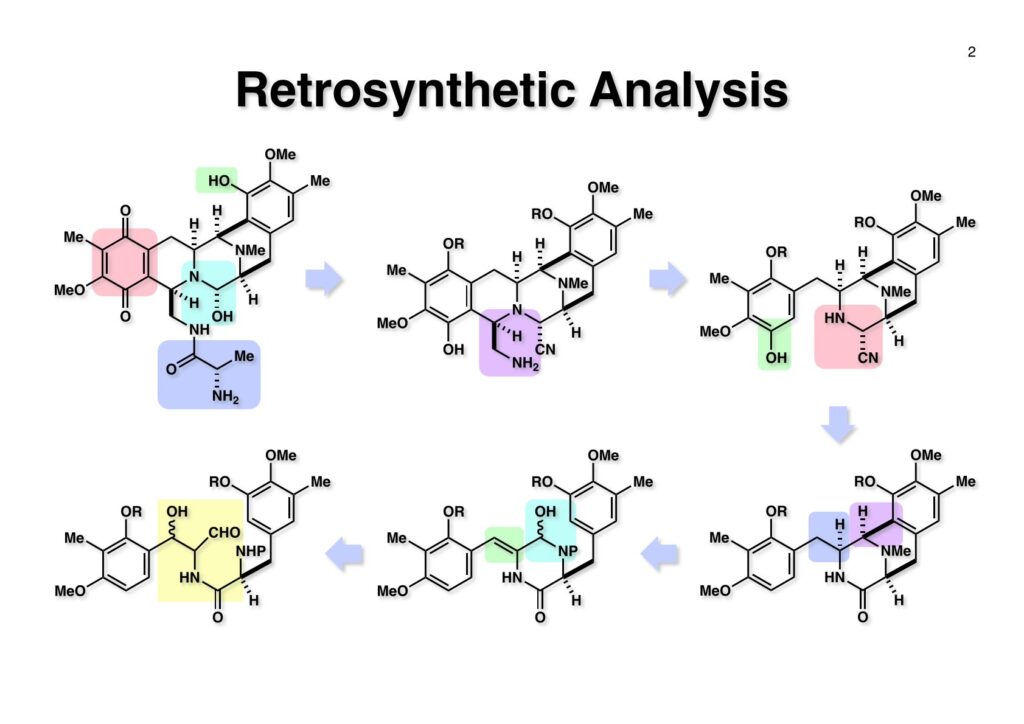
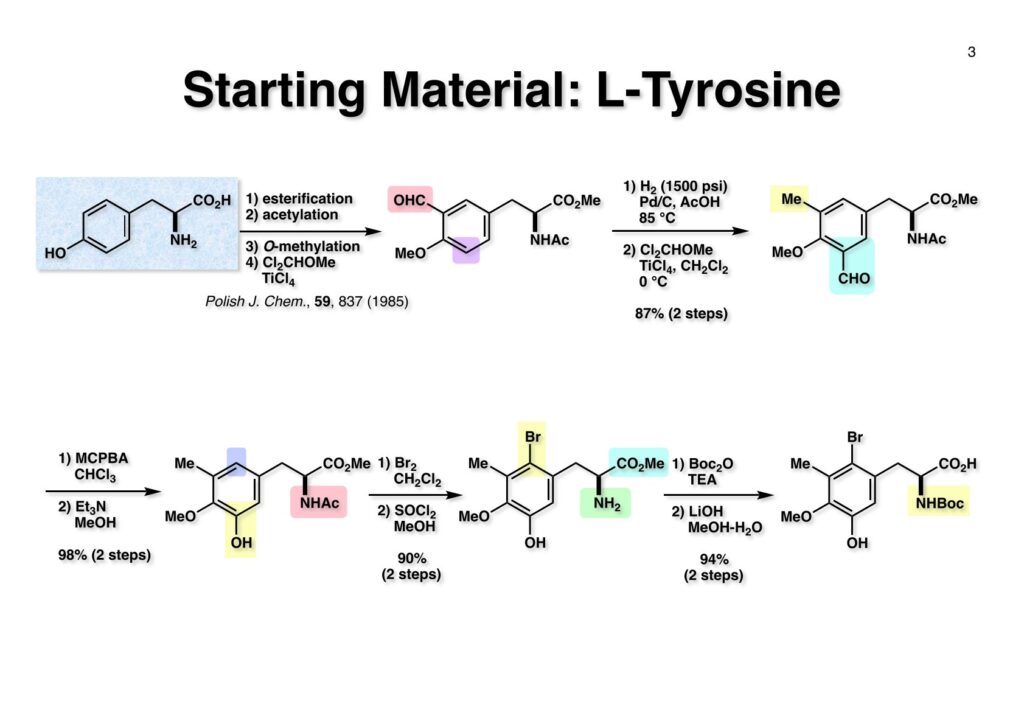
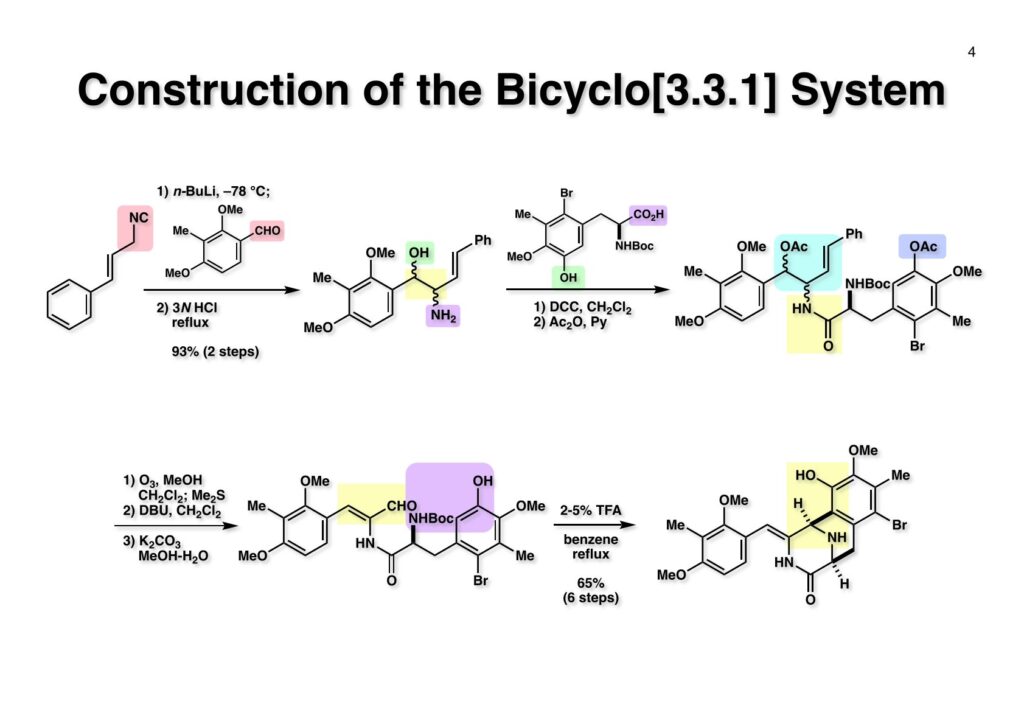
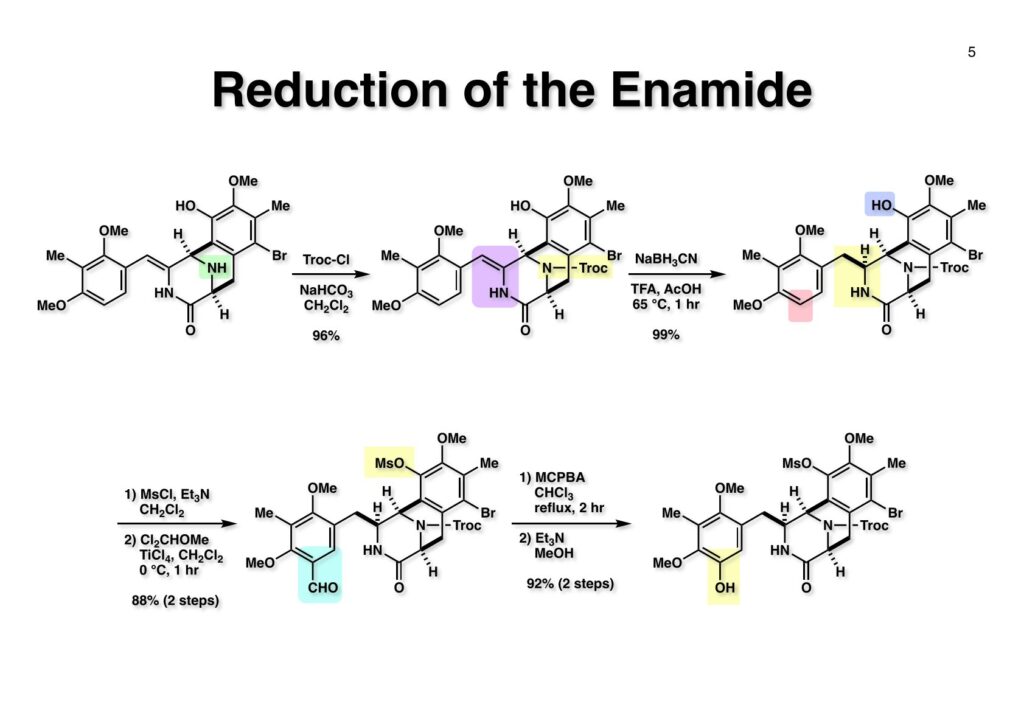
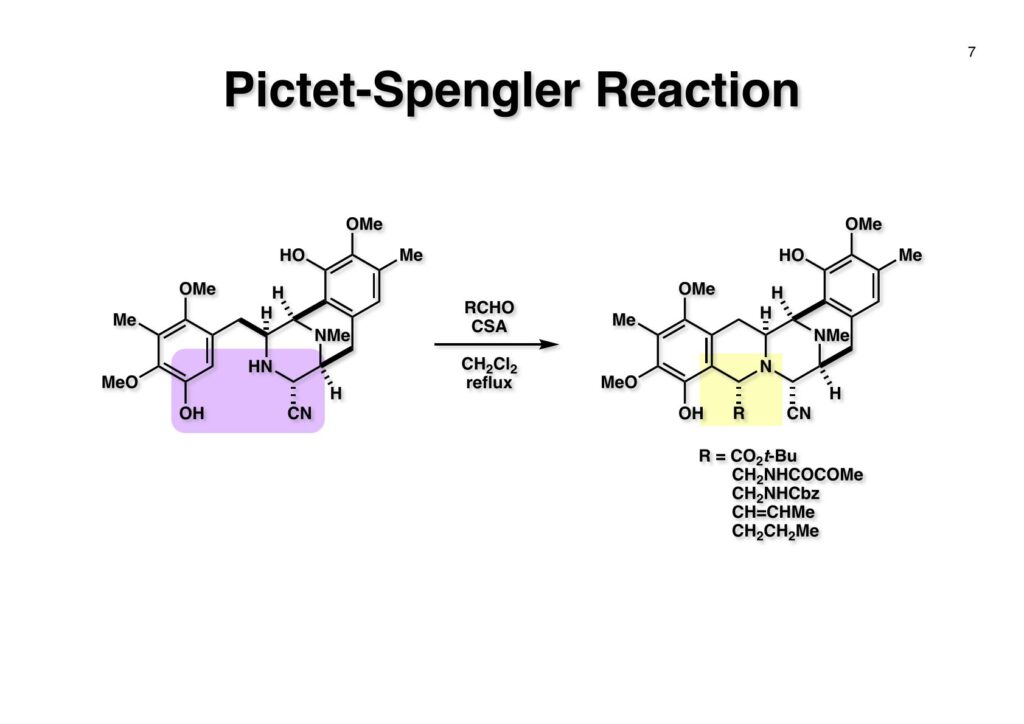
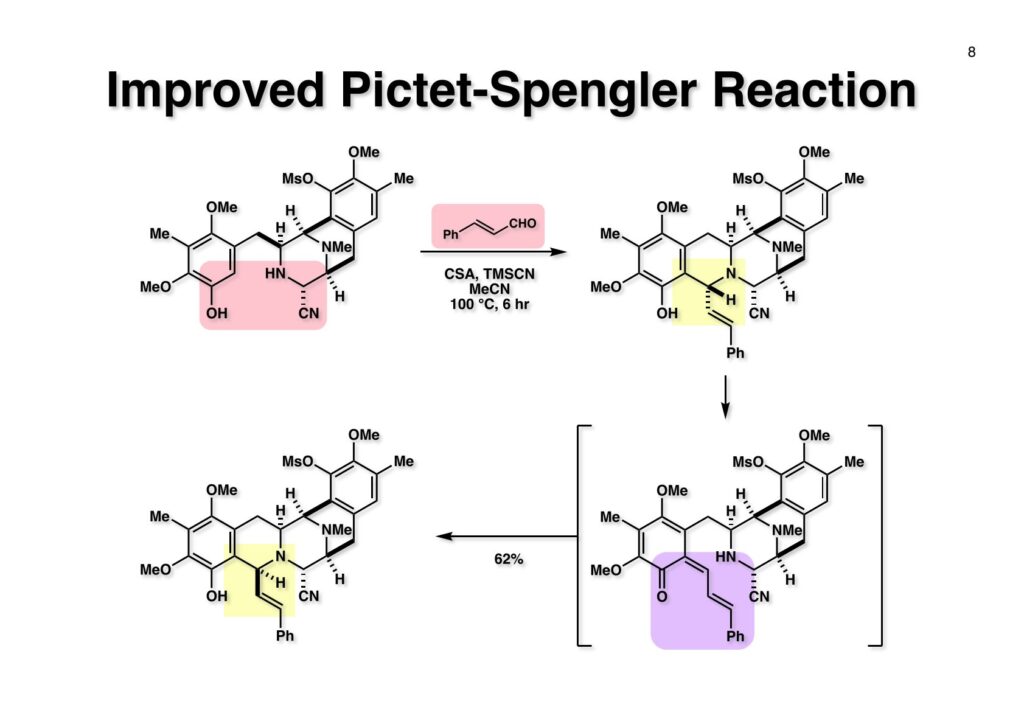
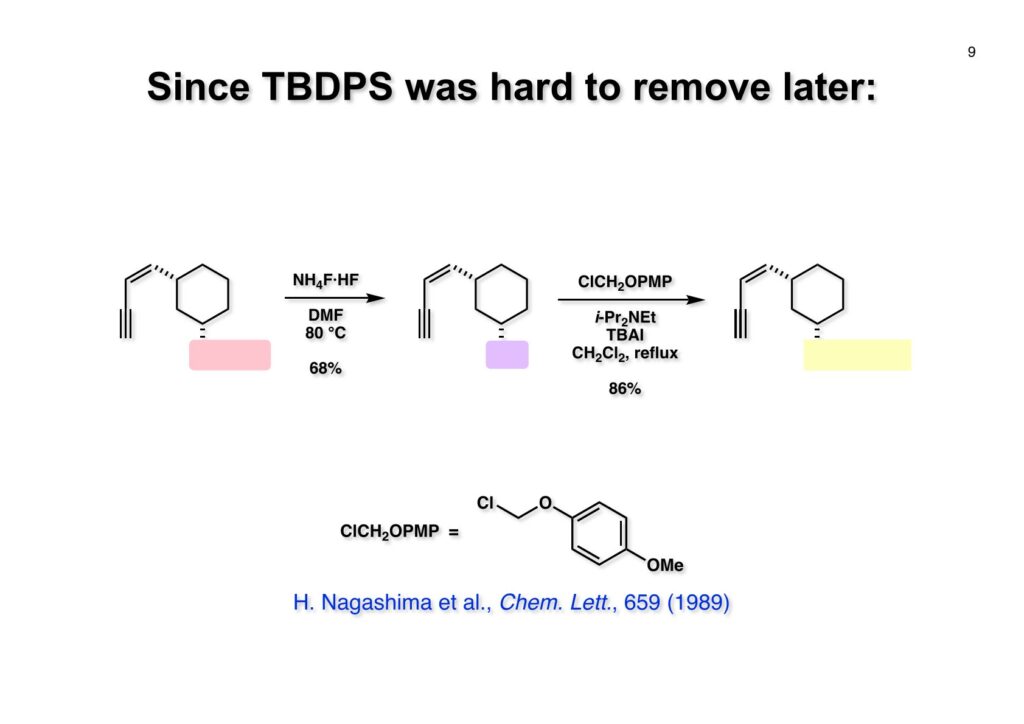
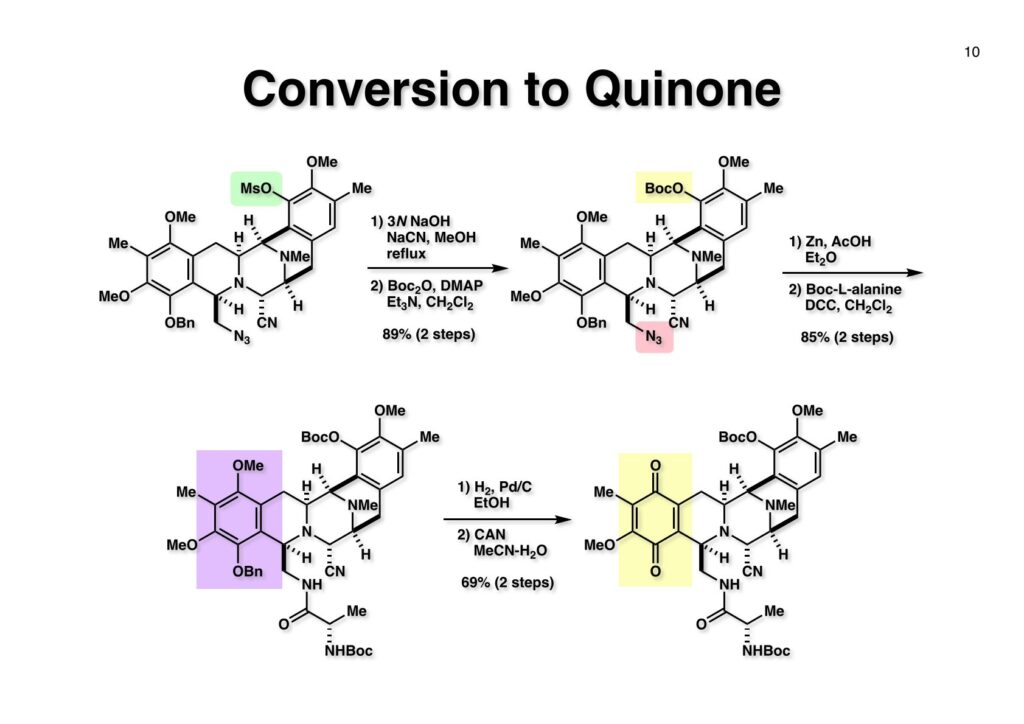
Safracin Bは1983年に吉富製薬(後に田辺三菱製薬に吸収合併され現在は田辺ファーマ)で単離構造決定された抗生物質で抗腫瘍活性も示すが薬にはなっていない。Saframycin Aに近い構造を有しており、ecteinascidin 743の合成ルート開発にも寄与すると思い、中国生まれで香港育ちのMui Cheungに全合成を始めてもらった。確かFlorida A&M University卒で、それほどアカデミック界では名の知られたところではなかったので、なんでわざわざ香港の高校からそこに留学したのか聞いた事があった。彼女曰く、ここなら3年で卒業できると思ったから、というのが記憶に残っている(実際3年で卒業してライス大学の大学院に入学)。頭も良く、腕も良い優秀な学生だったが、ソフトボールはやった事が無かったのか、私とキャッチボールをしたらグローブでなく顔で受けて?しばらく顔面に黒いアザが残ってしまった(そのうち消えたけど)。私がライス大学から東大に移る時に同僚のMarco Ciufoliniに彼女の面倒を見てもらうことにした。日本に到着した後にMuiから論文の原稿が送られてきたが、かなり直さないといけないと思いつつ、後回し、後回しにしていたらもう論文を出すのが面倒くさくなってそのまま今日に至ったという、彼女の顔をまともに見られない為体である。Muiはさらにthiangazoleというtantazole系の化合物の全合成も達成したが、基本的にはtantazole Bの全合成ルートを踏襲しているので、まあ論文を出すほどでもないか、とボツにしてしまった。こんな酷いボスの下で可哀想なMuiはよく頑張ったな、と思う。ただ、彼女はノシル基の開発に関しては大きな貢献をしたので、まあ勘弁してくれと、か細い声でささやいている。なおMuiは現在GSK (GlaxoSmithKline) Pharmaceutical CompanyのVice President, Global Discovery Chemistryとなって米国の製薬業界で活躍しているので遠巻きながら応援している。(この項を書き終えてからMuiに謝罪のメールを送ったところ明るく許してもらったことを記しておきます)
Mui Cheung, Ph.D. Dissertation, Rice University, 1997. (The total synthesis of safracin B was completed in 1994.) ChungKuang Jow made a great contribution to this synthesis. I would like to take this opportunity to apologize deeply to Drs. Mui Cheung and ChungKuang Jow for not publishing this interesting work.