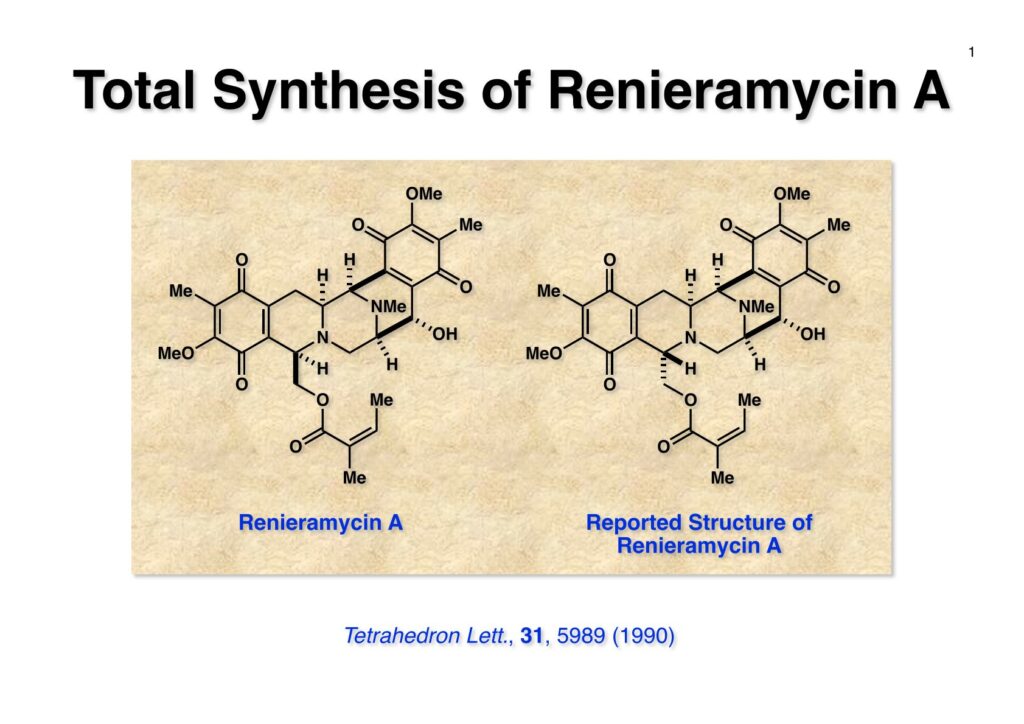
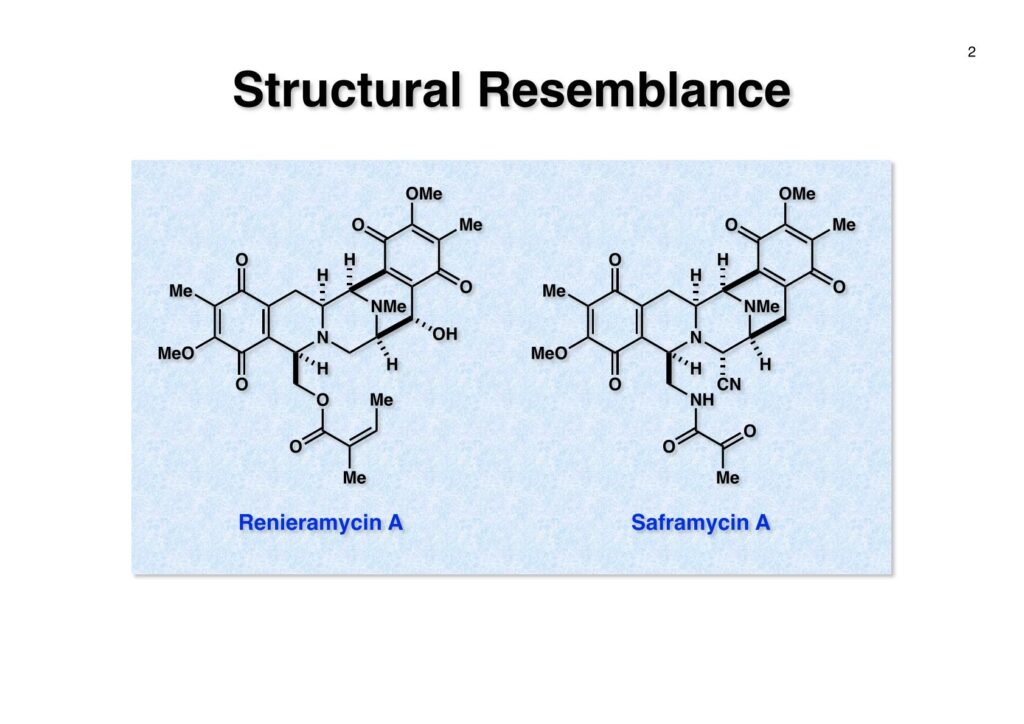
Renieramycin AはScripps Institution of OceanographyのJohn Faulkner教授が海面から単離構造決定した化合物でsaframycin類と構造がよく似ていたが、側鎖の立体化学がsaframycinとは逆であった。丁度saframycin Aの全合成をやっていたので、院生のMin Min Tunに全合成を始めるように頼んだが、彼女は修士号を取得して出ていったので、同じく院生のSteve Lintonにバトンタッチして全合成完成に至った。途中でFaulknerの決定した側鎖の立体化学が違うことが判明したので、私たちの全合成の論文を出す前に彼に連絡して構造を修正した論文を出してもらった。
“Total Synthesis of (±)-Renieramycin A,” T. Fukuyama, S. D. Linton, and M. M. Tun, Tetrahedron Lett. , 31 , 5989-5902 (1990).
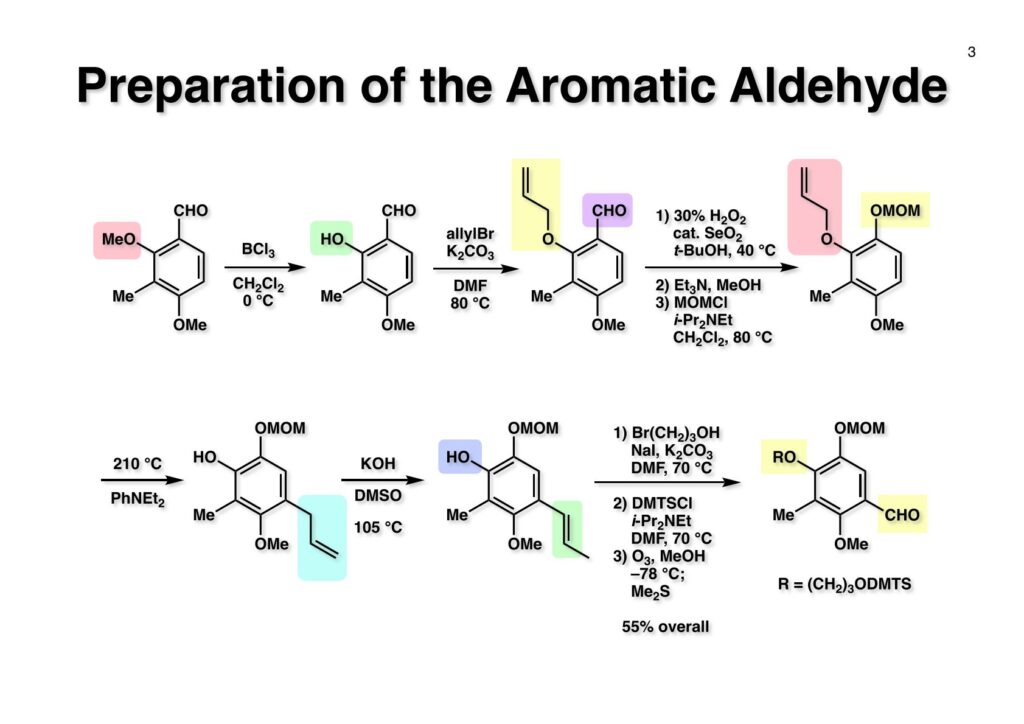
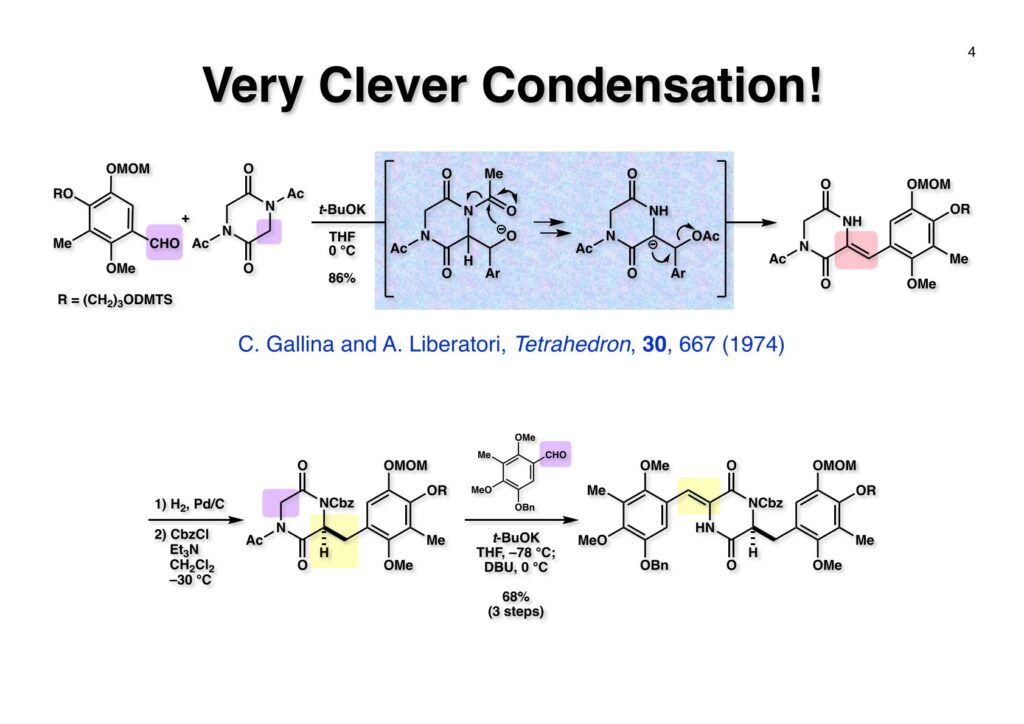
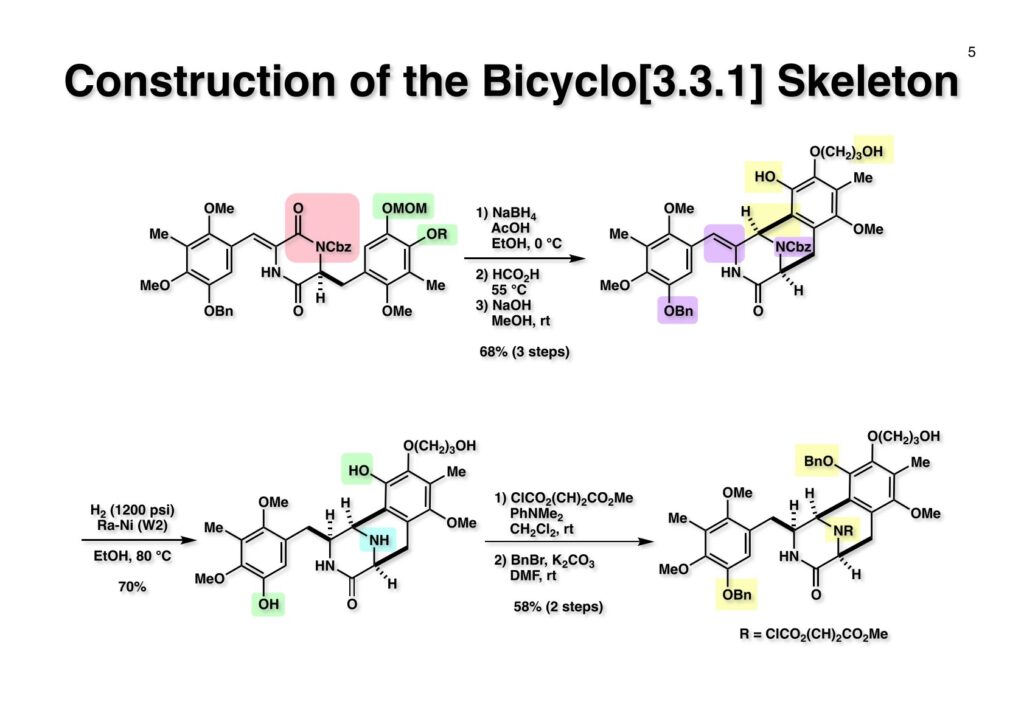
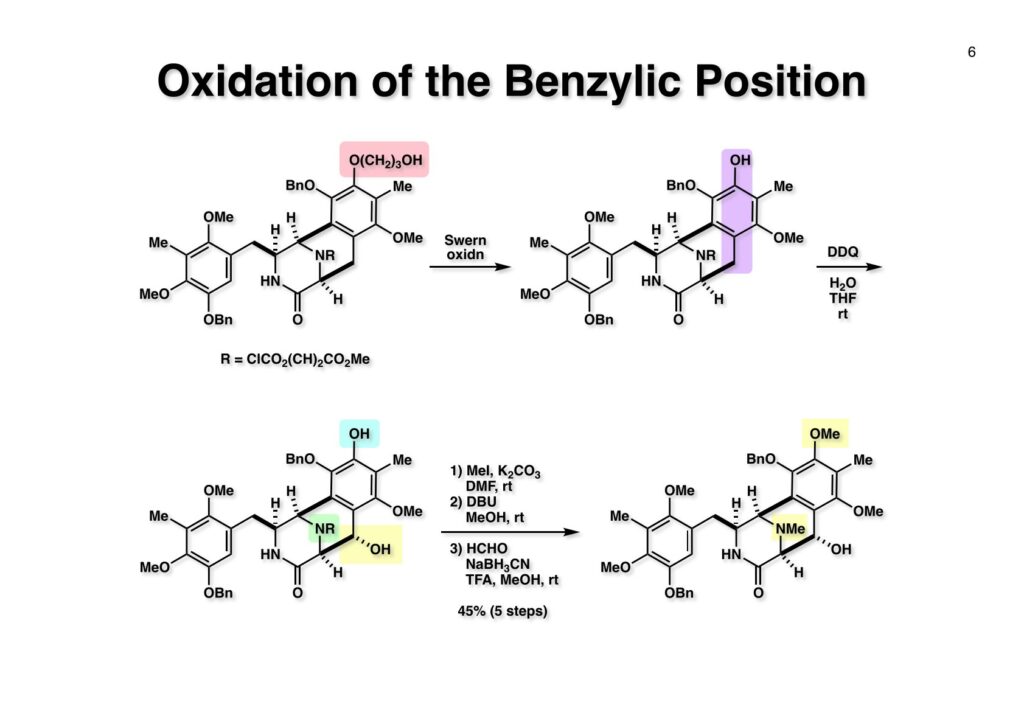
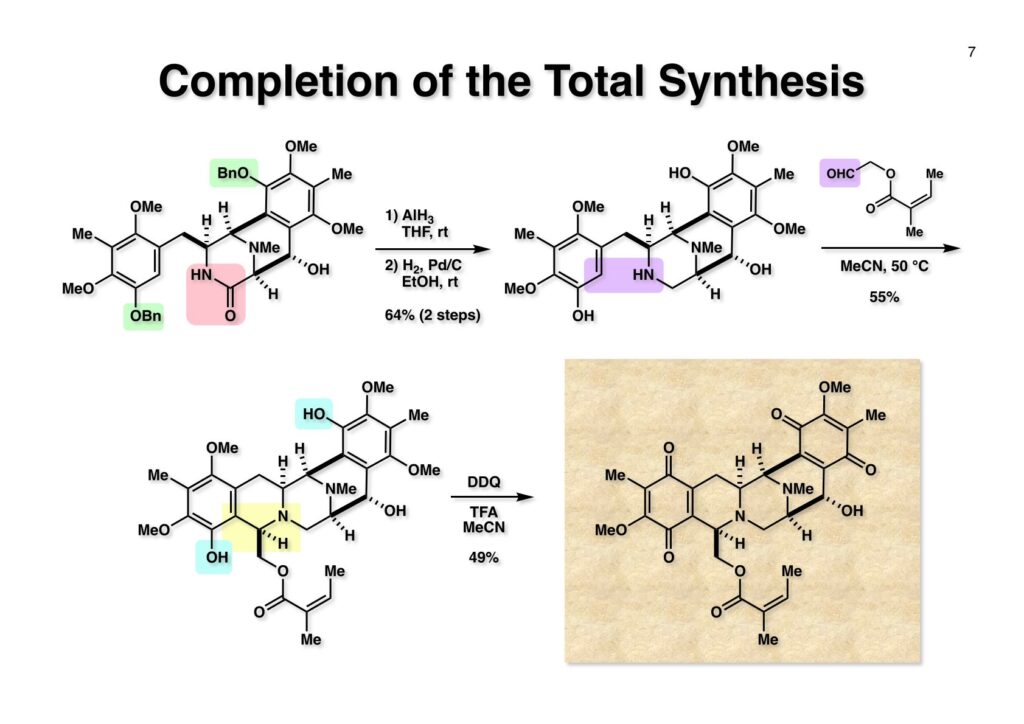
Renieramycin Aの全合成経路はsaframycin Aと類似しているので、ここでは逆合成解析は省略する。興味のある方は「saframycin A, Bの全合成」をご覧あれ。 (1-1) をBCl3と反応させるとカルボニル基のオルト位のMeO基だけが脱メチル化する。AlCl3を使っても同様の反応が起きるがこちらは反応速度が遅い。隣に配位できる官能基が無い場合はBBr3を使わないと実用レベルの脱メチル化は進行しない。ただし、オルトジメトキシベンゼンの場合はBCl3を使うと一つだけ脱メチル化することが知られている。得られた (1-2) のフェノールをアリル化して (1-3) を得た。次に触媒量の二酸化セレン存在下に30% H2O2をt -BuOH中で40 °Cを越えない速度で滴下するとBaeyer-Villiger酸化が進行してフェノールのギ酸エステルが得られる。この反応はオルト位かパラ位に電子供与基が存在しないとカルボン酸への酸化が起きるだけである。反応液中でHOSe(O)OOHというMCPBAのような過酸が生じて酸化が進行すると思われる。反応温度が高くなるとギ酸エステルが加水分解されてフェノールとなり、さらに酸化が進んでキノンが複製するために反応温度を低く保つ必要がある。なぜMCPBAを使わなかったかは大量スケールでの後処理の分液操作が圧倒的に簡単であるからだ。次にメタノール中でEt3Nを加えてギ酸エステルを加メタノール分解し、生じたフェノールをMOM化して (1-4) が得られた。フェノールのギ酸エステルはこの条件で簡単に加メタノール分解を受け、溶媒を留去すれば次の反応にすぐ使えるので便利である(NaOHを使うと分液処理が必要になるので)。アリルエーテル (1-4) はPhNEt2中で210 °Cに加熱することでClaisen転位が進行してパラ位にアリル基が入った (2-1) が得られる。オルト位が空いていればもちろんオルト位への転位が優先するが、オルト位にBr基やCHO基が存在する場合はそれらが脱離してオルト位にアリル基が入る場合があるので注意が必要である。次に (2-1) をDMSO中でKOHと加熱することで二重結合がベンゼン環と共役した (2-2) を得た。(2-2) の水酸基を3-bromopropanolでアルキル化し、さらに水酸基をDMTS (dimethylthexylsilyl) 基で保護した後にオゾノリシスを行ってアルデヒド (2-3) を通算収率55%で得た。 GallinaとLiberatori (Centro di Studio per la Chimica del Fannaco del CNR, Istituto di Chimica Farmaceutica dell’Università, 00185 Roma, Italy)が報告したdiacetyldiketopiperazineとアルデヒドとの縮合反応はニッチ的ながら非常に有用な反応である。(1-1) と (1-2) のTHF溶液にt -BuOKのt -BuOH溶液を加えていくと、イミドのアセチル基側ではなくメチレン基側が脱プロトン化されてアルデヒドに付加した (1-3) が生成する。アルコキシアニオンは近傍のアセチル基を攻撃してアセチル基が転位した (1-4) が生じる。ここでイミドのメチン基が脱プロトン化されてアセテートが脱離するが、嵩高い芳香環はカルボニル基を避けるようにZ -配置をとった化合物 (1-5) を与える。残ったメチレン基はイミドではないのでt -BuOKによる脱プロトン化は起きず、2個目のアルデヒドが縮合することはない。次に(1-5) の二重結合を接触還元し、ラクタムのNHにCbz基を導入した (2-1) を得た。ここで不斉還元ができれば光学活性体の合成が簡単になるのだが、この時点で不斉還元を試みたことはない。うまくいけば面白いのだが。 (1-1) のイミドカルボニル基をNaBH4-AcOHで還元し、ギ酸中55 °Cで加熱するとacyliminium ionが生じてbicycle[3.3.1]骨格が形成されるDMTS基はこの酸性条件で外れてしまい、1級アルコールはギ酸エステルになってしまうのでアルカリ加水分解して (1-2) を得た。(1-2) の接触還元はかなり抵抗し、Raney-Ni (W2)を触媒として高温 (80 °C) 高圧下 (1200 psi) で行って (2-1) が得られた。次にClCO2(CH2)CO2Meでアミンを保護してからフェノールのベンジル下によって (2-2) が得られた。 (1-1) のヒドロキシプロピル基はSwern酸化の条件で直接フェノール (1-2) を与えた。ここで含水THF中でDDQ酸化を行うとフェノールのパラ位のメチレン基が酸化されて水がビシクロ系のexo 側から付加した (2-2) のみが得られた。この酸化反応はアミンをメチル化した状態ではうまく進行しなかったのでウレタンとして保護しておいた。(2-1) のフェノールをメチル化後にDBUを用いてアミン保護基をretro-Michael反応で除去し、還元的メチル化によって (2-2) を得た。 ラクタム (1-1) の還元はAlH3を用いた。以前にも述べたが、アミド(ラクタム)の還元はLiAlH4よりはAlH3の方が良い結果を与える事が多いと記憶している。LiAlH4とAlCl3を計算量混ぜて作っていた(3LiAlH4 + AlCl3 = 4AlH3 + 3LiCl)。次にベンジル基を加水素分解して (1-2) を得た。アルデヒド (1-3) を用いたPictet-Spengler反応はアセトニトリル中50 °Cで行い、5:1の比で望むβ体とエピマーのα体が生成し、主生成物 (2-1) をクロマトで分離した。最後に (2-1) をDDQ酸化することでビスキノン体であるrenieramycin A (2-2) の全合成を完了した。