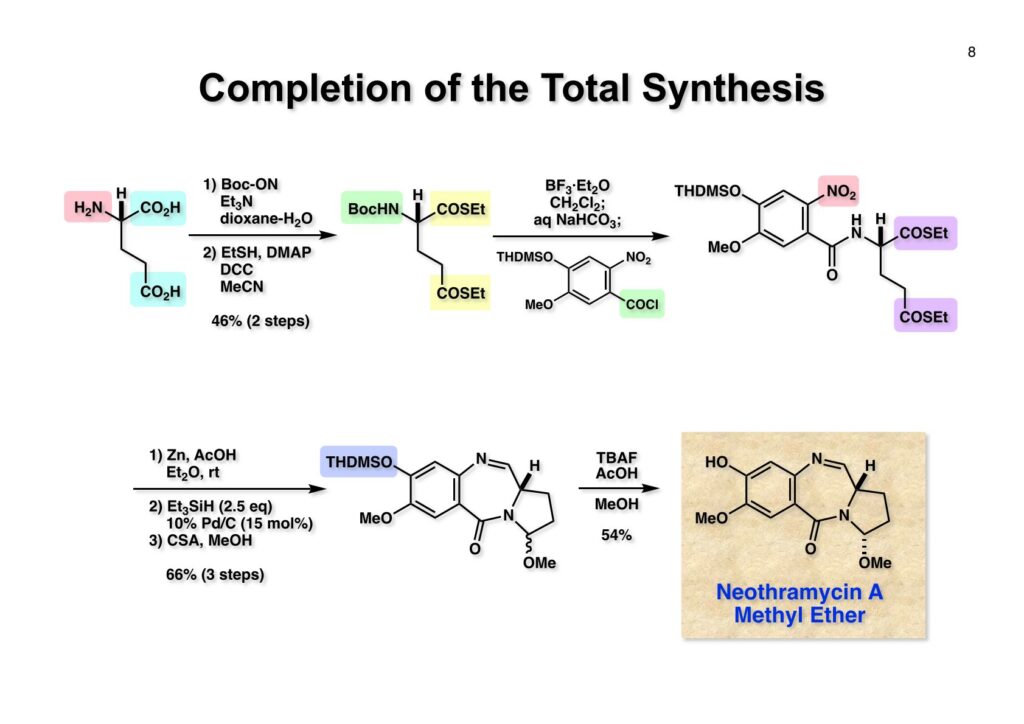
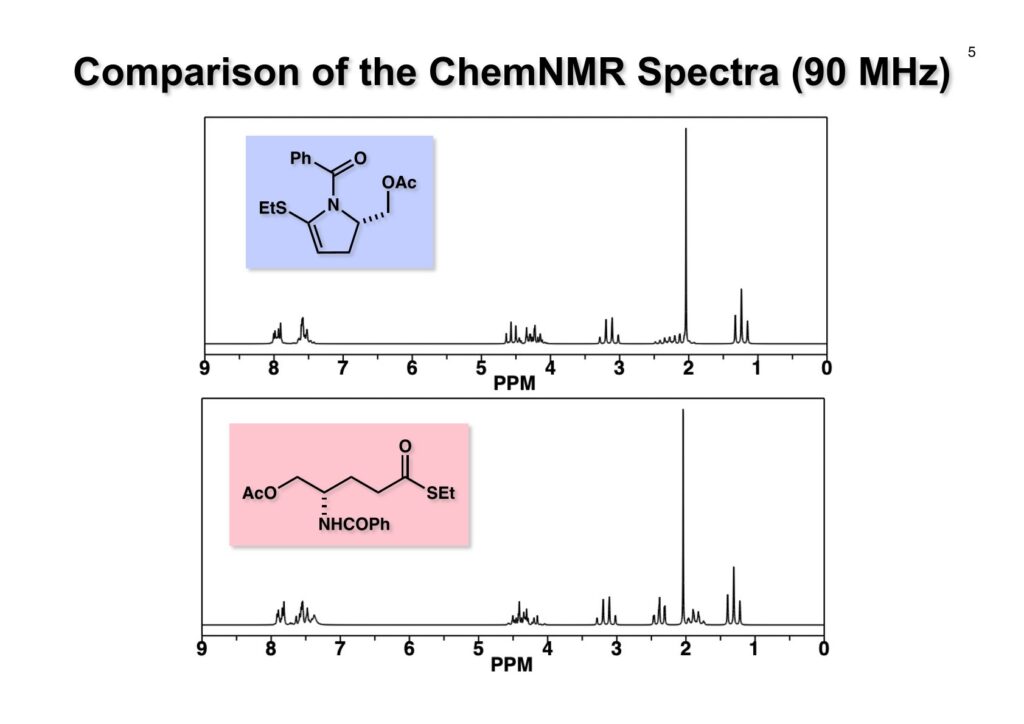
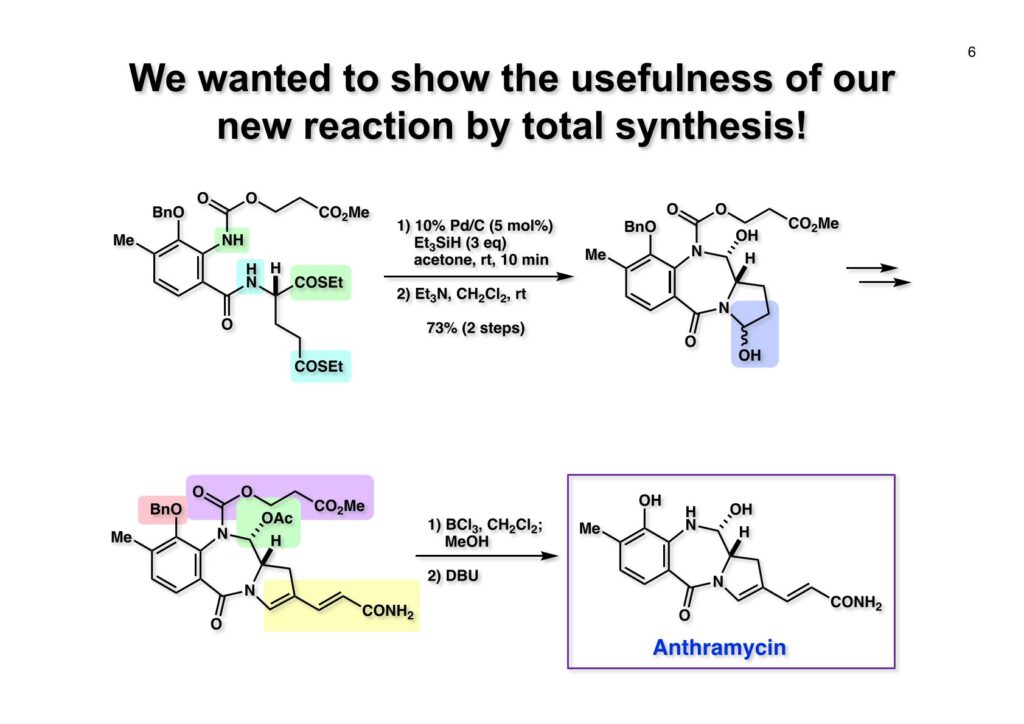
Neothramycin自身は不安定でメチルエーテルとして単離した全合成であるが、これは本来の全合成標的化合物ではなかった。John Linという確かお父さんが台湾出身のライス大学卒の院生で、アメリカでは大学を卒業して同じ大学の大学院に進学するのは少数で他大学の大学院に進学するのが普通だ。彼は多分自宅から通いたかったのだと思う。非常に真面目な学生で私のくだらない冗談は分かってくれるが、自分から冗談を言うのは聞いたことがない。ほんの少しだけLeping Liに助けてもらったが基本的にはJohnの研究で、本来はanthramycinの全合成に挑んでいた。Anthramycin全合成の最後まで辿り着いたのだが、ラセミ化しているかどうかが判明せず、途中で発見したチオエステルをアルデヒドに変換する反応を世に出すのを先行させたので、anthramycinは傍に置いて、eothramycinの全合成に適用して新反応を発表した。Anthramycinの全合成は結局棚に放置したままで埃を被り、結局興味が無くなってJohnの博士論文に収めるだけに終わった。
“Facile Reduction of Ethyl Thiol Esters to Aldehydes: Application to a Total Synthesis of (+)-Neothramycin A Methyl Ether,” T. Fukuyama, S.-C. Lin, and L. Li, J. Am. Chem. Soc., 112, 7050-7051 (1990).

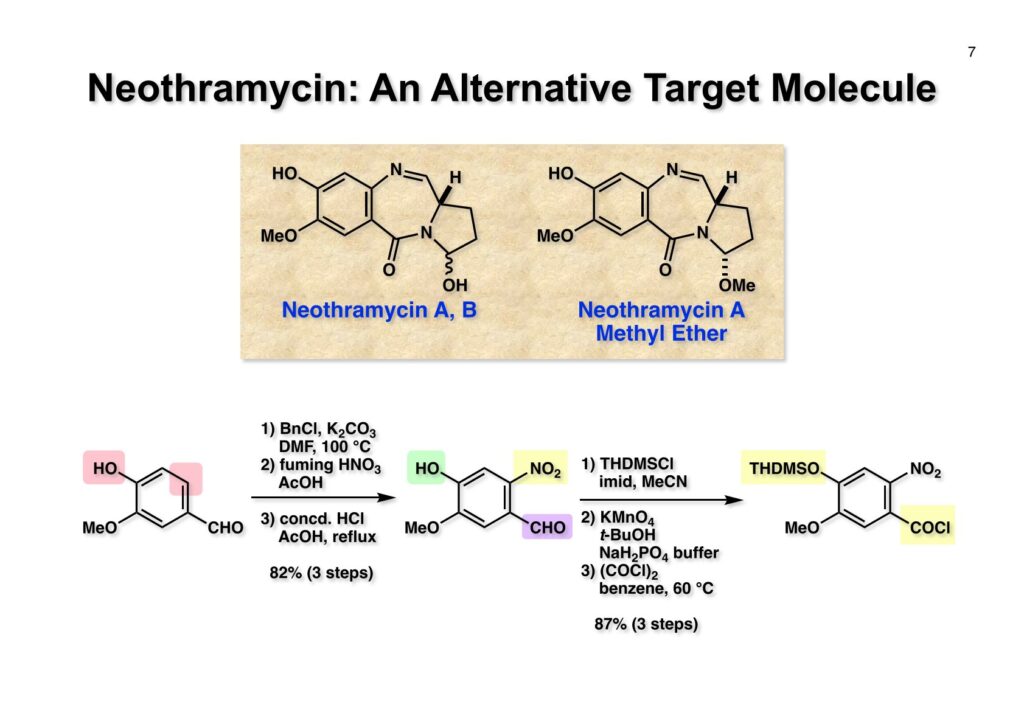
市販のバニリン (2-1) のフェノールをベンジル基で保護してからMeO基のパラ位をニトロ化し、次に酸性条件でベンジル基を外して (2-2) を得た。次にフェノールをシリル基(THDMS = thexyldimethylsilyl) で保護し、過マンガン酸カリでアルデヒドをカルボン酸に酸化した。カルボン酸はoxalyl chlorideを用いて酸塩化物 (2-3) に変換した。