三共から来た研究生の小林聡さんが中心となり、日本新薬からの研究生だった上田稔浩さんが加わって第一世代インドール合成法を使ったvindolineの全合成を青息吐息で完成させた。当時はvinblastineの全合成など「夢のまた夢」というのが正直なところだったが、catharanthineの全合成(いずれ解説する予定)途上で開発した第二世代インドール合成法が非常に有望だったのでvinblastineの完全全合成に挑戦することにした。Gelsemineの全合成を達成した横島聡君をメインに窪山剛之君と佐藤綾人君が主としてvindolineの改良合成に従事した、当研究室としては珍しく多人数の努力の結晶として完成させたプロジェクトである。
“Stereocontrolled Total Synthesis of (+)-Vinblastine,” S. Yokoshima, T. Ueda, S. Kobayashi, A. Sato, T. Kuboyama, H. Tokuyama, and T. Fukuyama, J. Am. Chem. Soc. , 124 , 2137-2139 (2002).
“An Efficient Total Synthesis of (–)-Vindoline,” S. Kobayashi, T. Ueda, and T. Fukuyama,. Synlett, 883 (2000).
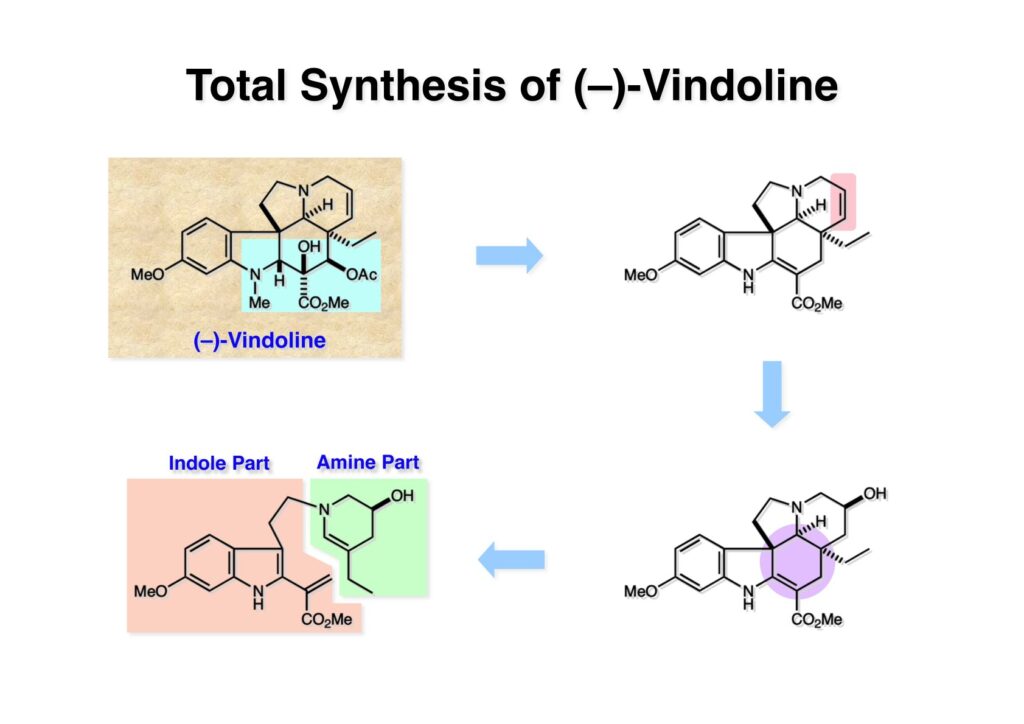
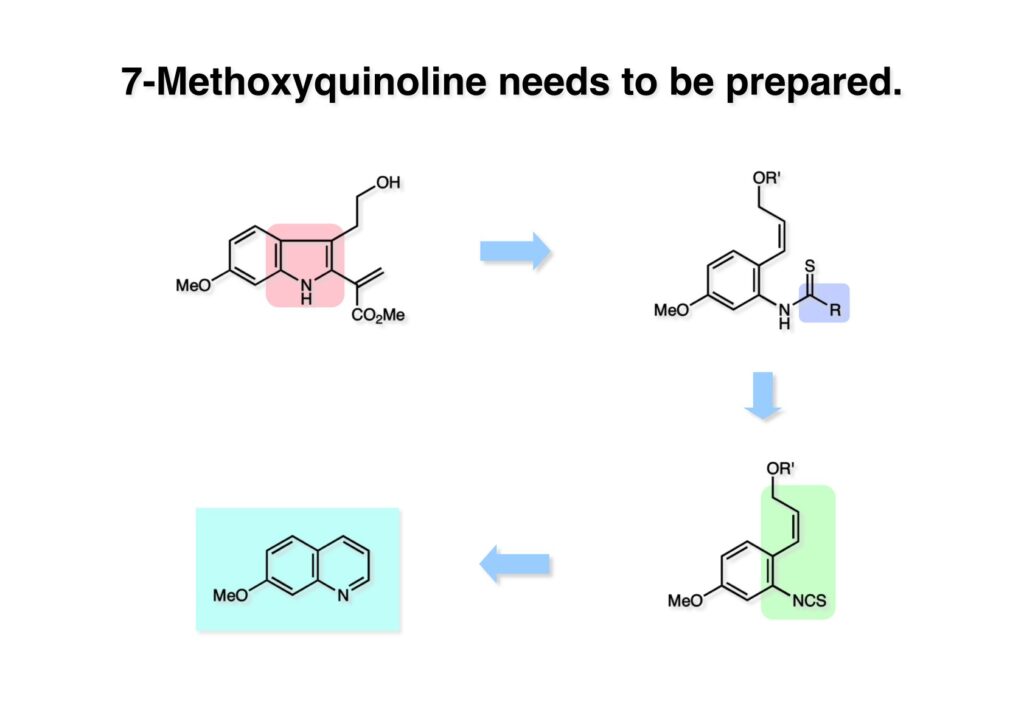
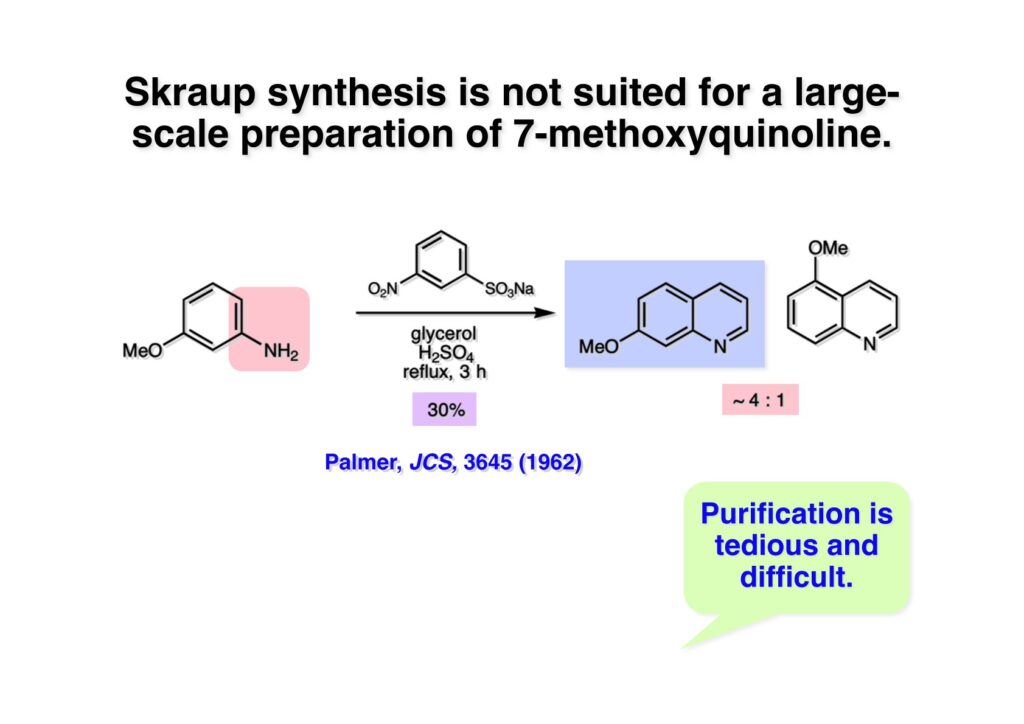
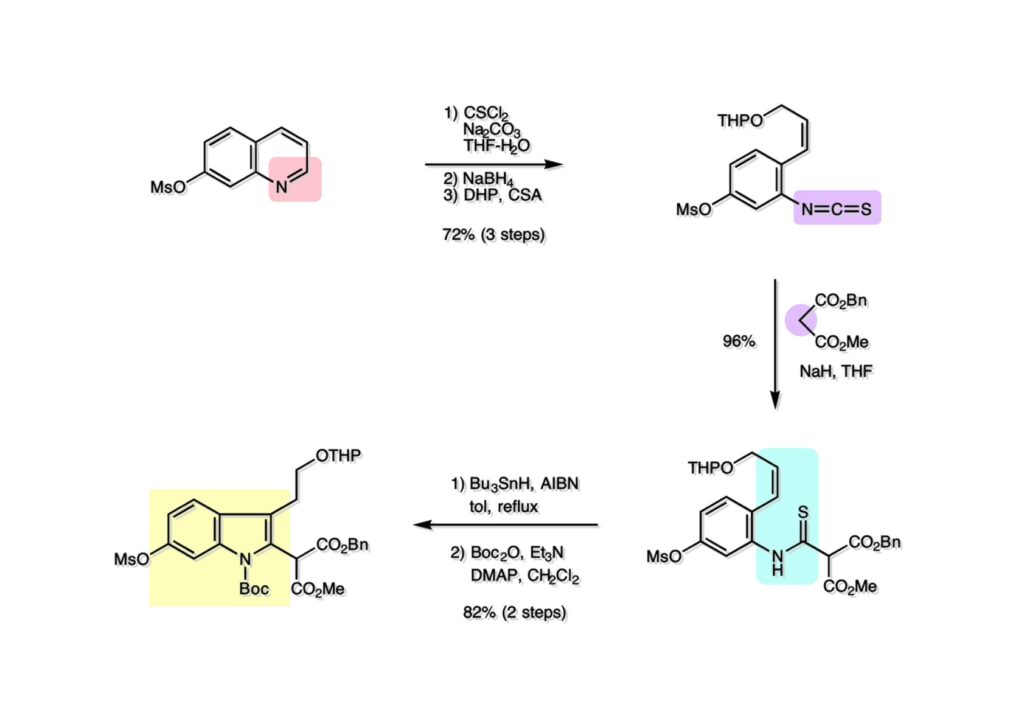
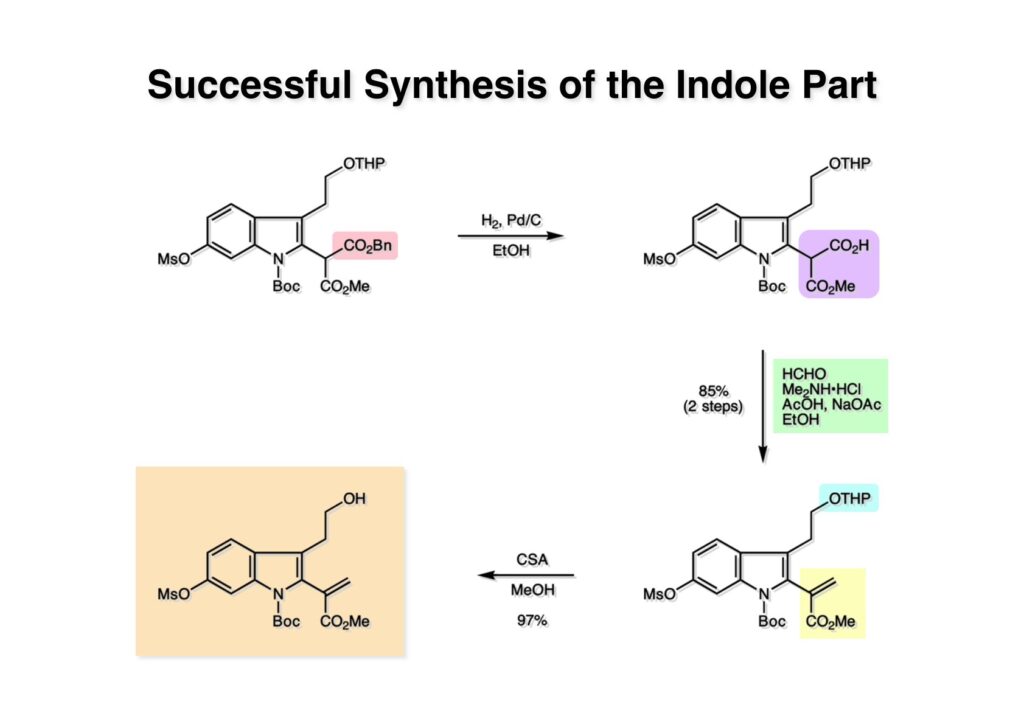
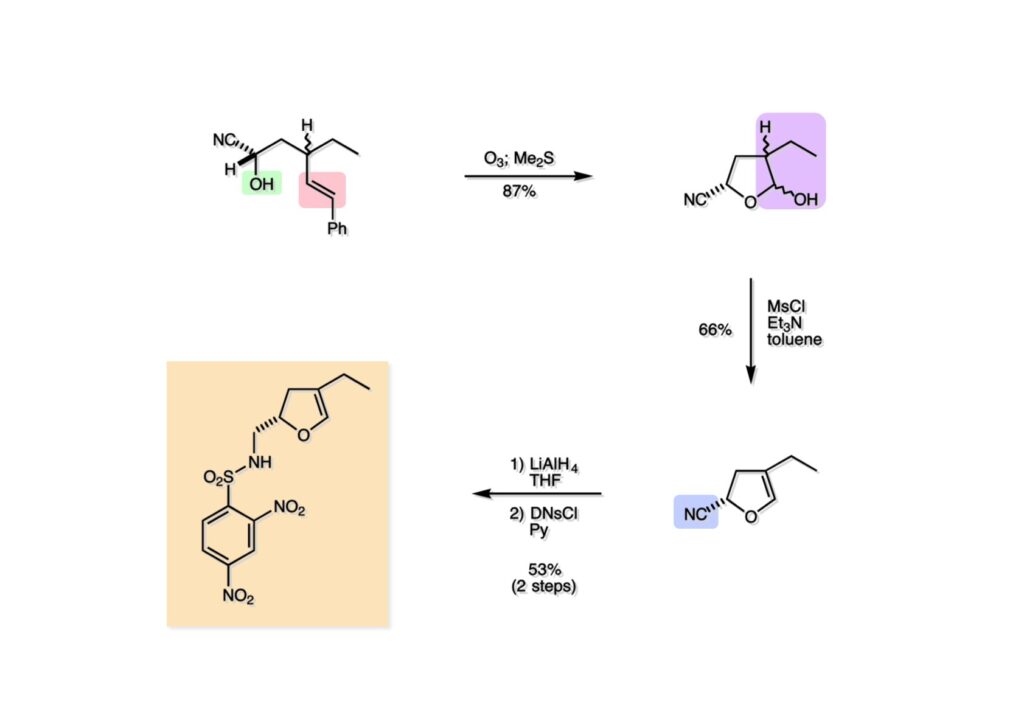
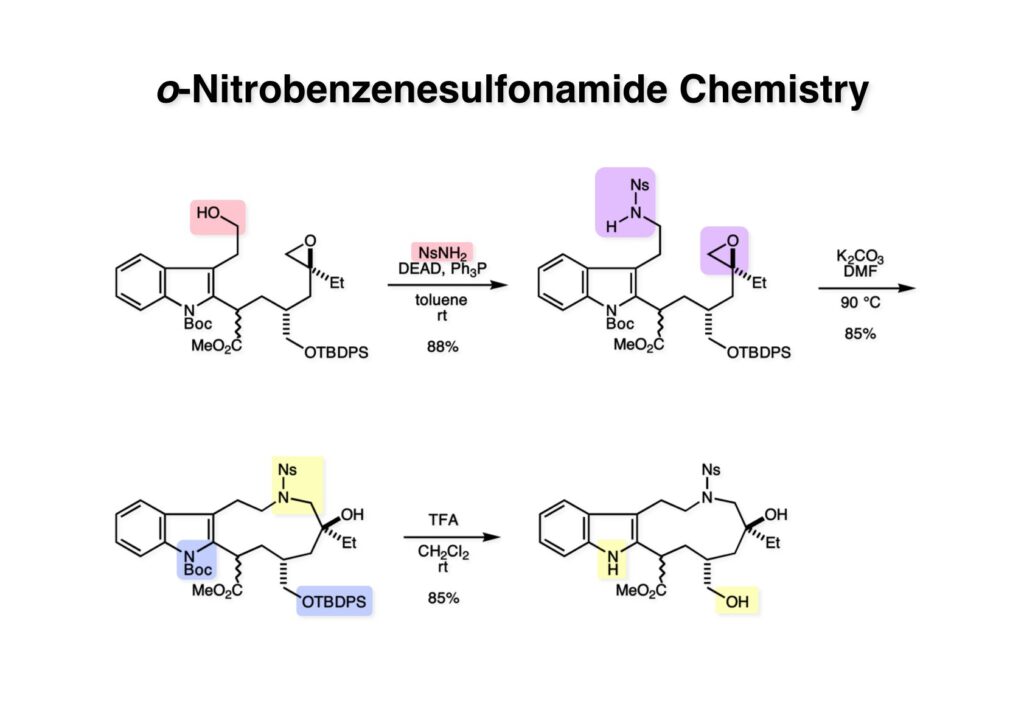
Vinblastine (1-1) は西洋ニチニチソウの葉から得られる抗がん剤で主として悪性リンパ腫などの治療に用いられている。私たち以前の合成は天然のvindoline (1-2) を用いているため、完全全合成は未達成だった。チオアミド (2-1) を出発物とした高効率的な第二世代インドール合成法を開発したので、vindolineを全合成的に利用可能なスケールで合成できる目処が立ち、vinblastineの全合成を開始した。 1987年にVermont大学のMartin Kuehne教授が中間体 (2-1) を経由した(-)-vindoline (1-1) の全合成を報告した。アミン部位の水酸基の立体化学が環化体 (2-2) の立体化学を制御するという画期的な研究で、私たちも重要中間体 (2-2) の合成に着手した。 (1-1) のようなインドールを合成するためにはチオアミド (1-2) が必要で、第二世代インドール合成法ではイソチオシアネート (2-2) からチオアミドを合成していた。(2-2) のようなイソチオシアネートはキノリン (2-1) にチオホスゲンを作用させることで合成できる。そこで7-methoxyquinoline (2-1) の大量合成を行うことにした。 キノリンの合成法と言えばクラシカルなSkraup合成が有名であるが10グラムのm -アニシジン (1-1) とグリセリンを原料に常法で行ったところ7-methoxyquinoline (1-2) と4-methoxyquinoline (1-3) の4:1の混合物が得られた。反応液は真っ黒で3リットルの分液ロートに移したが水層と有機層の境目が全く分からず、チョロチョロと水層を流して、流れが変わったところが有機層なんていう酷い目にあった。何とかクロマト分離をして目的物を3グラムほど得たが、とてもスケールアップする気にはなれない反応だった。というわけで、自前でキノリン合成を開発しなければならない事になった。 3-aminophenol (1-1) をTsCl-Py で反応させるとアミンがトシル化された (1-2) が得られる。これをEt3Nに代えるとフェノールがトシル化される。更に注意しなければならないのはアニリンをTsCl-Et3Nの条件でトシル化すると二つトシル基が入ることがよくある。一つにするにはこれをアルカリ加水分解する必要がある。次に (1-2) をメタノール中でアクロレインとEt3Nで反応させるとMichael付加が起きて (1-3) になる。(1-3) をTHF中で3N HClと加熱するとフェノールのパラ位に環化脱水反応が起きてジヒドロキノリン (2-3) が得られた。これをDMSO中KOHと130度に加熱すると7-hydroxyquinoline (2-2) が得られる。この反応はTsアニオンが脱離するのか、Tsアミドが加水分解され、生じたジヒドロキノリンが空気酸化されてキノリンになるのかはハッキリしない。おそらく前者であると私は思うのだが。(2-2) の水酸基をメシル化して (2-1) に変換した。最終的にはこのキノリンを100グラムほど合成した。 キノリン (1-1) を水存在下でチオホスゲンと反応させるとReissert型の反応が進行してcis -α,β-不飽和アルデヒドをオルト位に有するイソチオシアネートが得られる。この反応はLancaster catalogという英国の試薬会社のカタログの中に収録されていたが、1992年頃のライス大学でのグループミーティングで学生たちに反応機構を考えさせたのが記憶に残っていた。ある程度性能の良い記憶装置を持つというのも「良いケミスト」の条件である(エヘン!)。アルデヒドとイソチオシアネートのどちらが反応性が高いのか知らなかったがNaBH4還元に付すとアルデヒドだけがアルコールに還元された。生じたアルコールをTHP基で保護することで (1-2) が得られた。このイソチオシアネートは様々なカルバニオンと反応してチオアミドを容易に与えた。ここではbenzyl methyl malonate (2-1) のアニオンと反応させて高収率でチオアミド (3-2) を得た。このチオアミドをBu3SnHとAIBNとともにトルエン中で環流するとBu3SnラジカルがチオアミドのSを攻撃し、生じた安定ラジカルが隣のオレフィンに付加することによりインドール環を形成することになる(第二世代福山インドール合成法)。インドールのNHをBoc化すると高収率で (3-1) が得られた。このインドール合成法は10グラムスケールでも問題なく進行したが、もっとスケールアップすることも容易だと思われる。 前ページでbenzyl methyl malonateを用いたのは勿論理由がある。片方のエステルだけを簡単にカルボン酸にするためだ。Methyl malonateの片方だけを大量に加水分解するには1当量のNaOHかKOHを使う方法が古くから知られている。片方がカルボキシレートになると電子吸引力が落ちるのでもう一つのエステルの反応性が低下するためだ。それは余談として、(1-1) を加水素分解してカルボン酸 (1-2) に変換し、Mannich反応を行うと脱炭酸とMe2NH脱離が同時に進行してアクリル酸誘導体 (2-2) が高収率で得られた。次にTHP基をメタノール中CSAで処理することにより除去してアルコール体 (2-1) を得た。 (1-1) のインドール部分の合成は計画通りに進行したが、アミン部分は光学活性体を合成しなければならない。ここではその逆合成解析を説明する。逆合成解析は起こりうる反応でエネルギーのより高い方向に進めるのが一般的だが、必ずしもそれに拘る必要はない。順方向に進める方法をいくつか知っているに越したことはないが、反応機構上無理で無さそうだったらどんどん進めて行って、ある時点で妥当かどうかを振り返り、面白そうなら実行に移すというようなザックリした考え方が良いと思う。逆合成解析の一般論を述べただけで、ここではそんな難しい解析は必要ない。まず、エナミン (1-2) を加水分解してアルデヒド (2-3) にすると水酸基を捕捉してラクトール (2-2) を構築できる。このラクトールには2つのキラル中心があるので簡略化のため脱水してジヒドロフラン環 (2-1) にしておけばよい。アミンの活性化基としては2,4-dinitorobenzenesulfonyl基を用いることにした。 前ページの (2-3) の逆合成経路は省略したが、アルデヒドは二重結合のオゾン分解、アミノアルコールはシアンヒドリンの還元、光学活性なシアンヒドリンはアセテートの酵素による加水分解をやろうと計画した。市販の2-pentenal (1-1) にPhMgBrを付加させてアリルアルコール (1-2) を得た。なぜPh基を付加させたかというと、続くClaisen転位の反応温度を下げるためだ。沸点が94度のbutyl vinyl ether (1-3) を溶媒にし、酢酸水銀によるoxymercurationを経由して (1-2) の水酸基をビニル化して環流温度で転位させる方法で大量(>100g)のアルデヒド (1-4) を合成した。(1-4) の酢酸溶液にNaCNを加えてシアンヒドリンにし、無水酢酸-Pyでアセチル化して (2-3) のジアステレオ混合物が得られた。この混合物をTHF-水中、50度でAmano lipase PSという酵素を用いると44%の収率 (97% ee) で目的とするシアンヒドリン (2-1) が得られた。未反応のアセテート (2-2) は加水分解して酸性にし、アセチル化することで (2-3) を再生することが容易にできた。
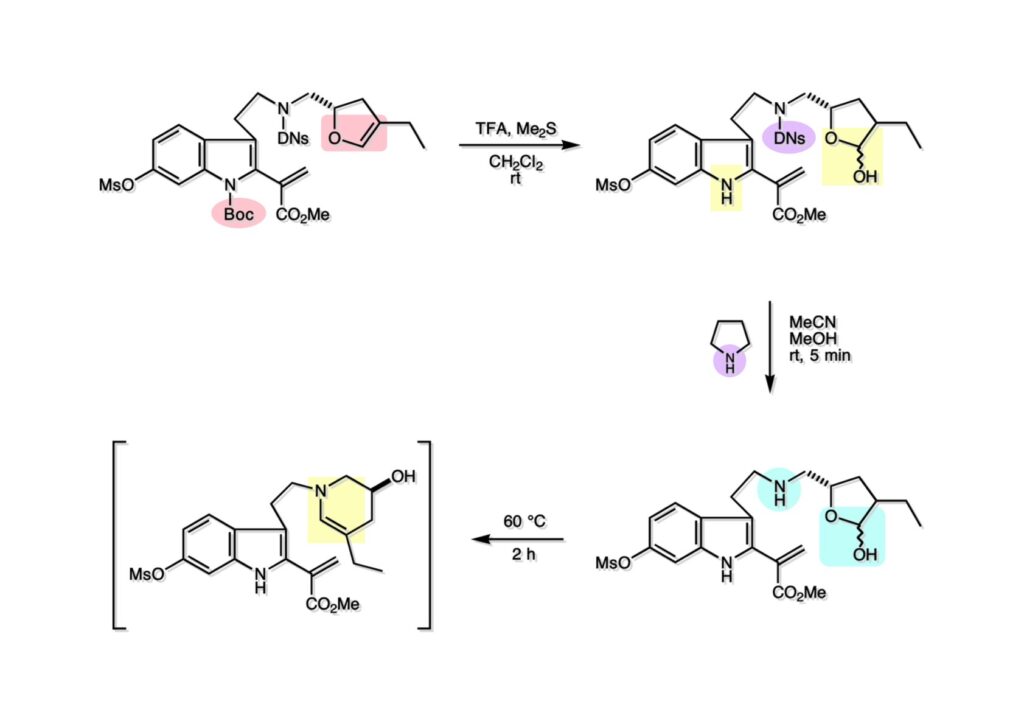
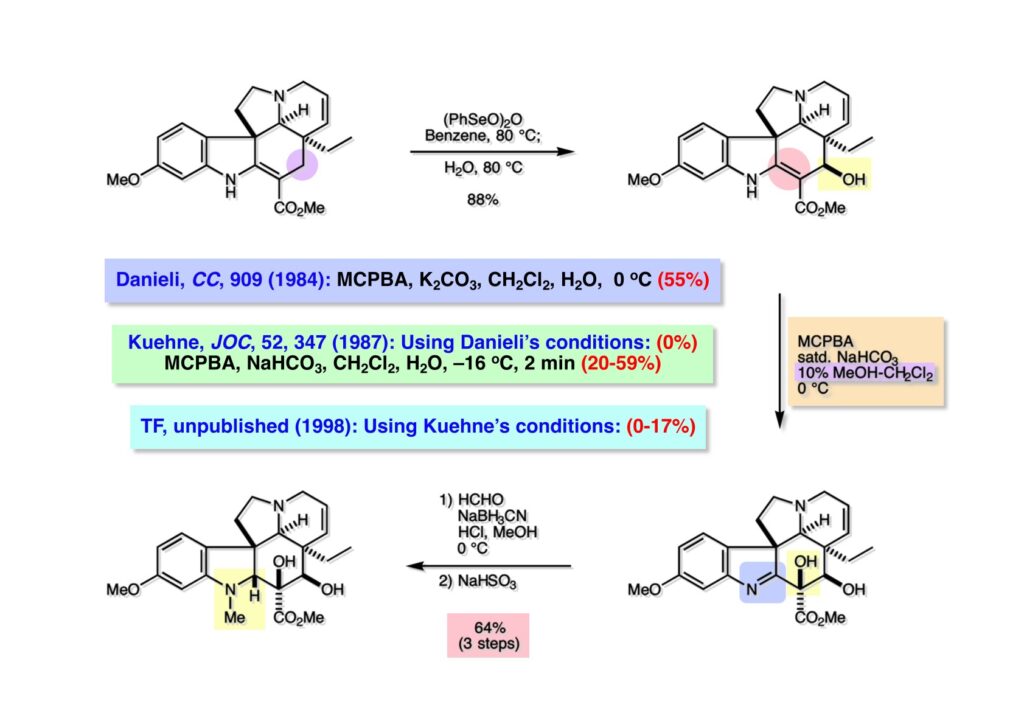
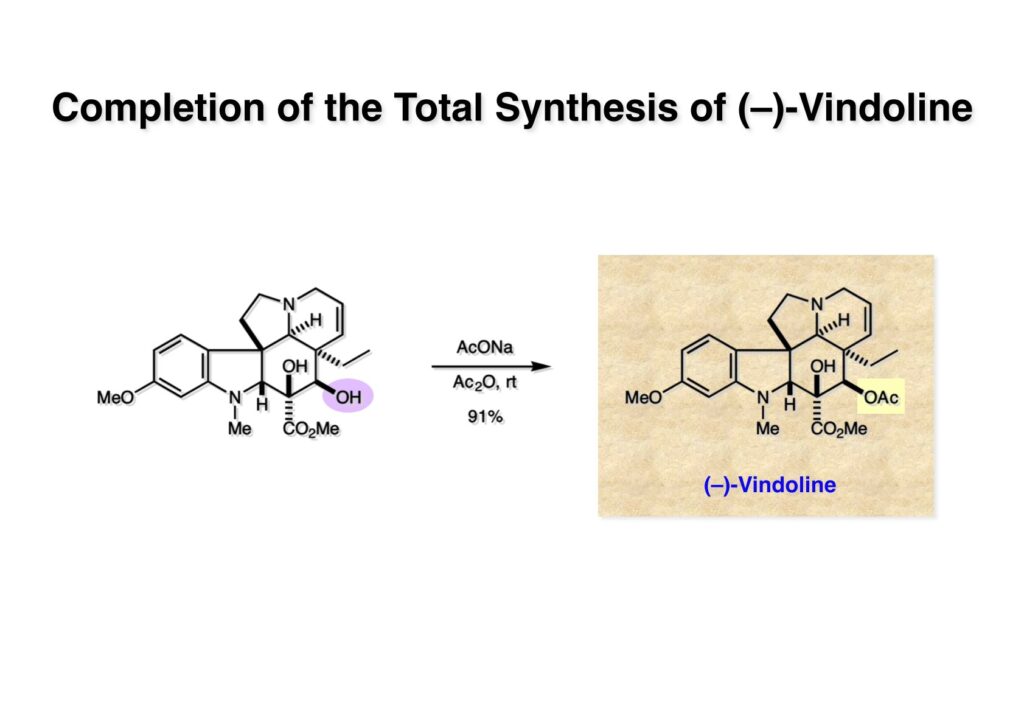
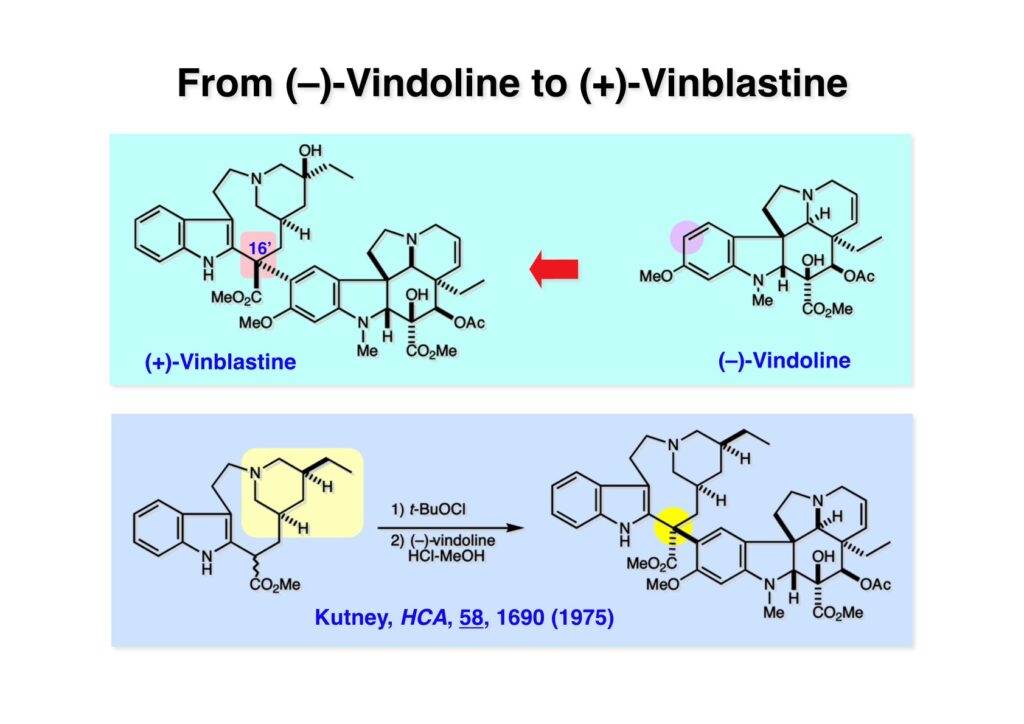
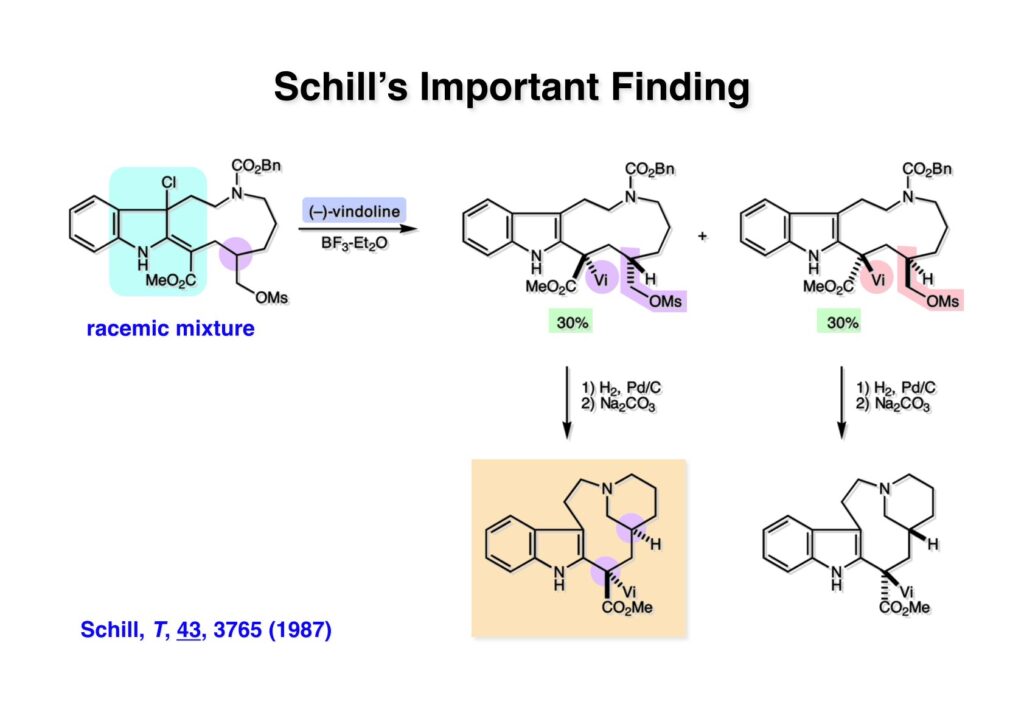
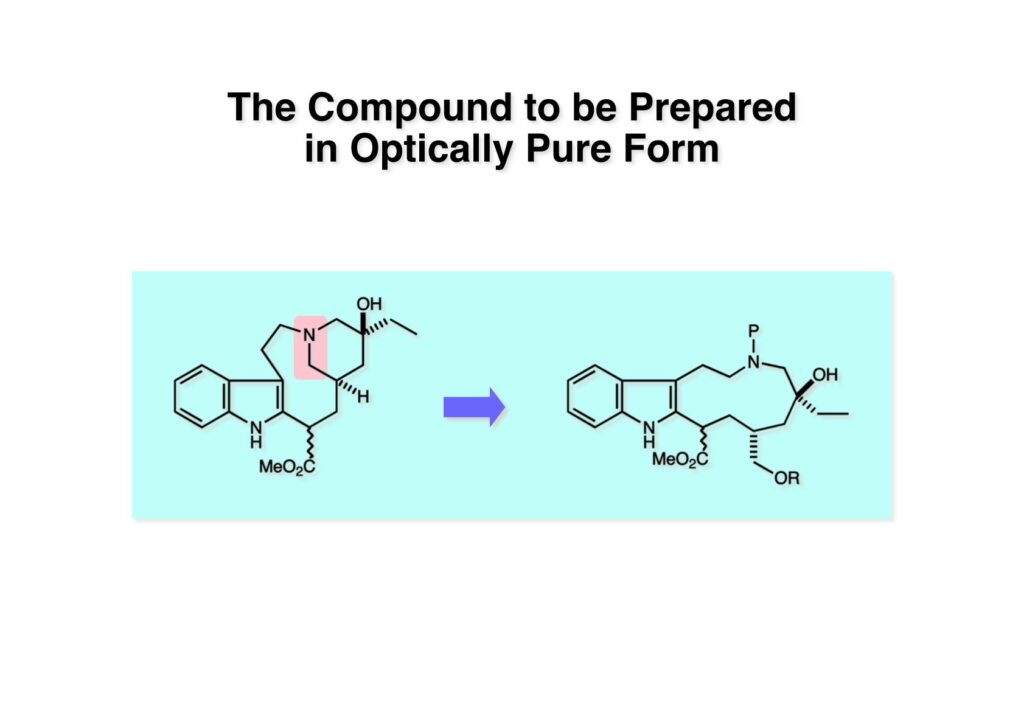
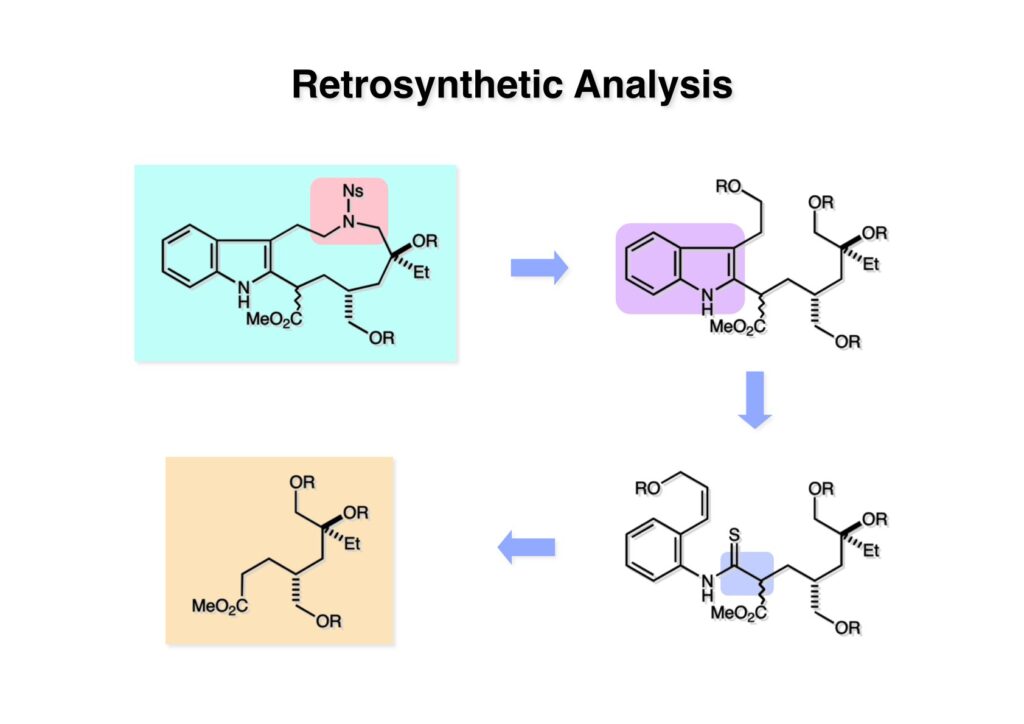
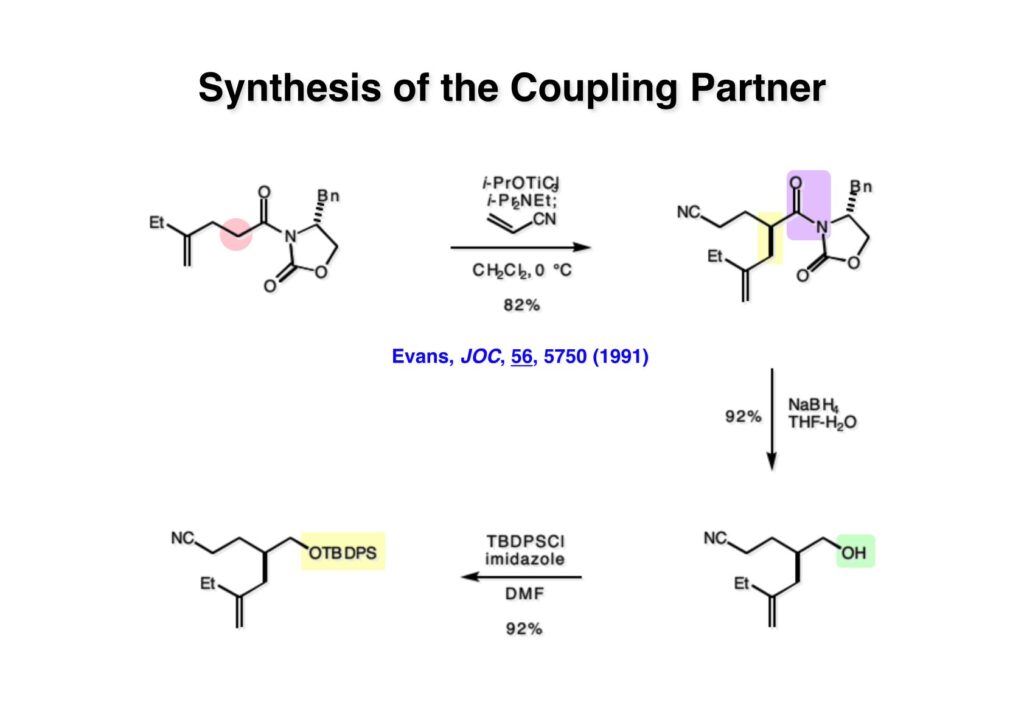
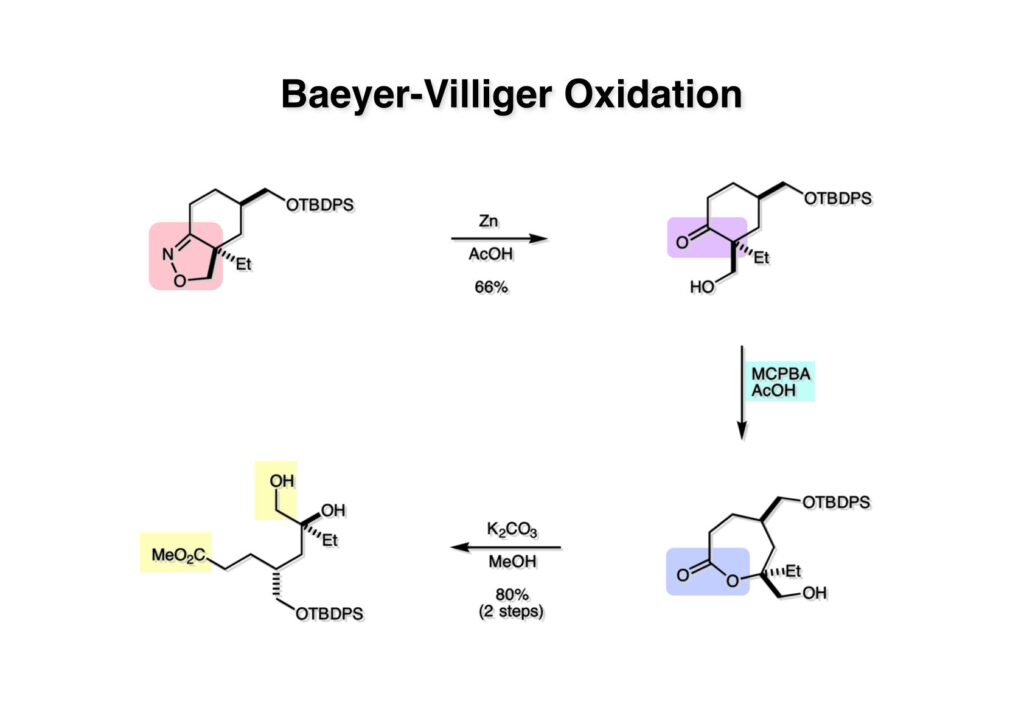
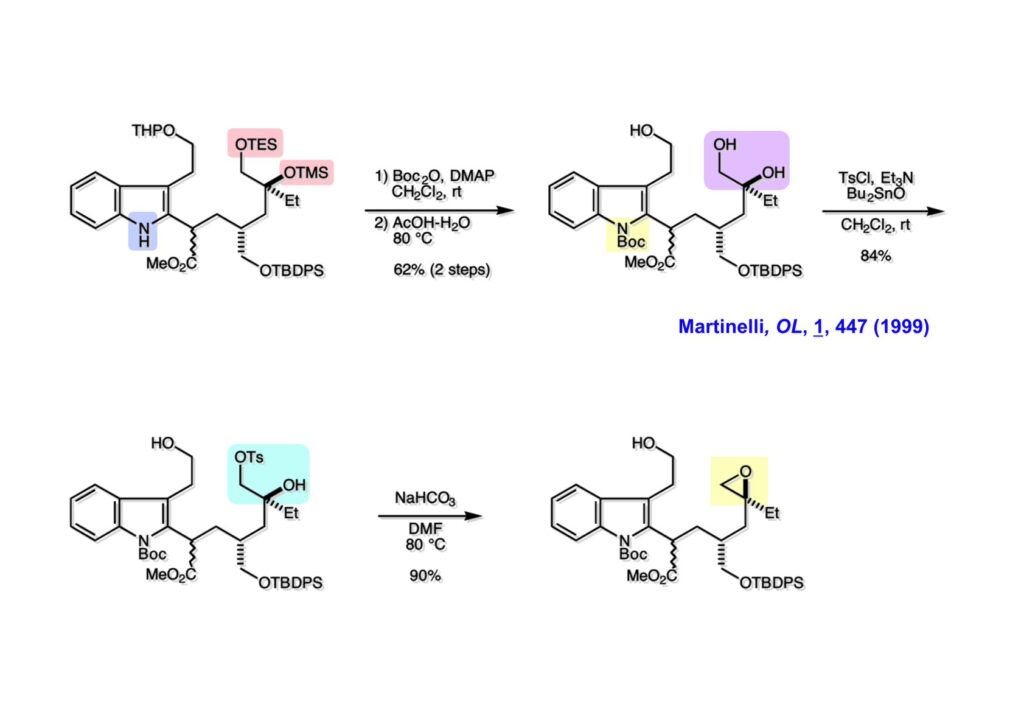
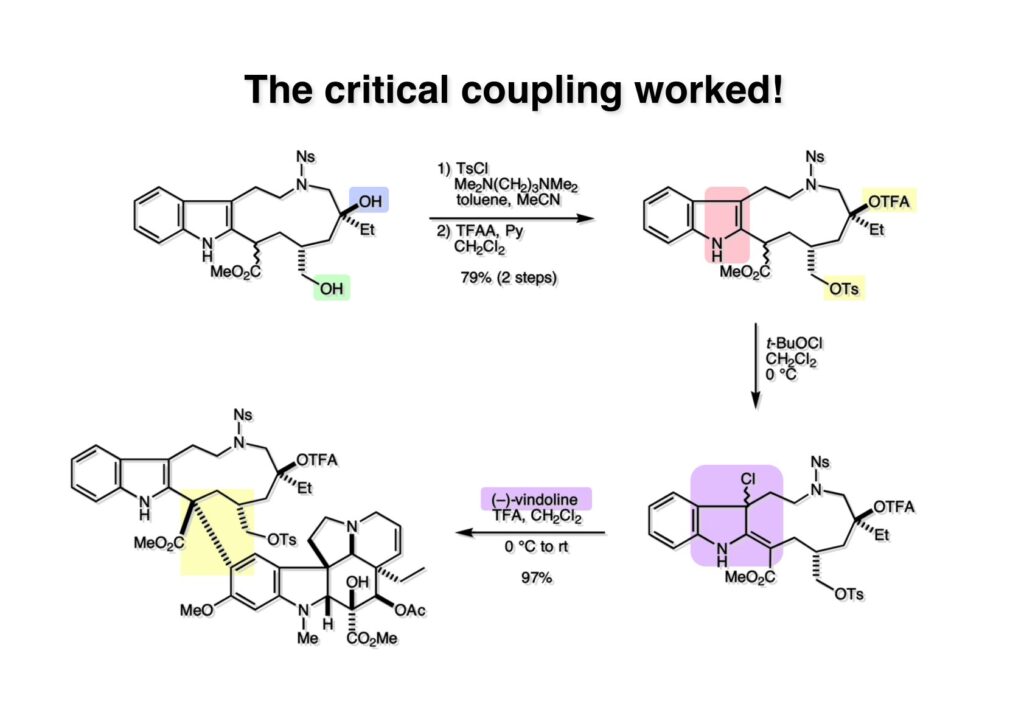
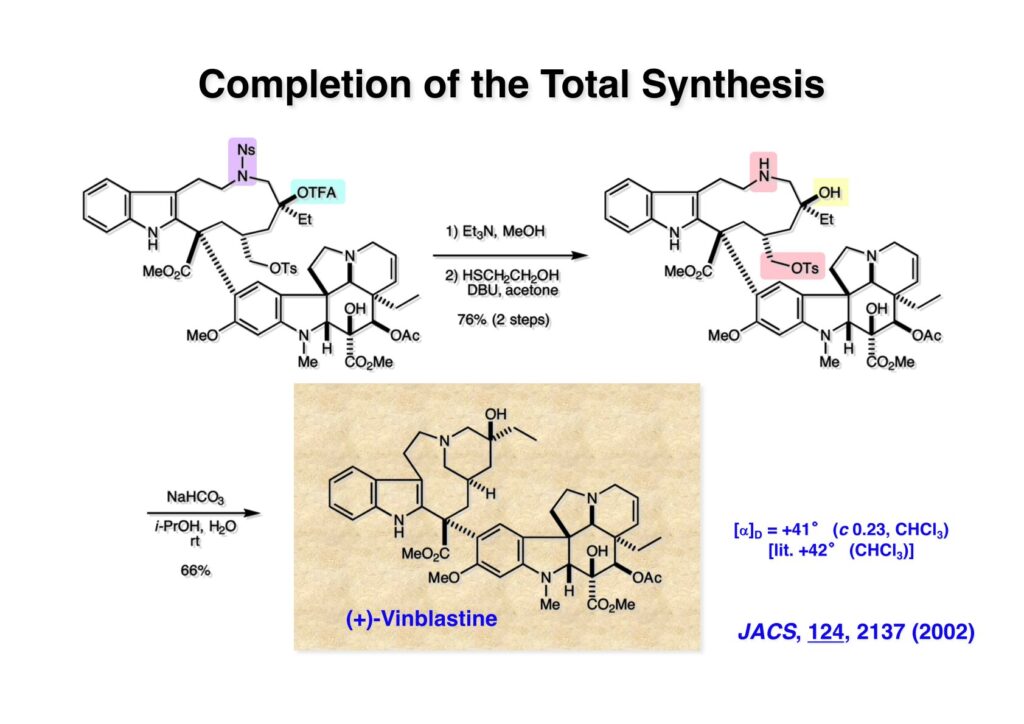
オレフィン (1-1) をオゾン分解してMe2Sを加えるとラクトールのジアステレオ混合物 (1-2) が得られた。これにトルエン中Et3N存在下でMsClを加えると容易にジヒドロフラン体 (2-2) を与えた。収率が多少低いのは揮発性が高く蒸留によって化合物を得たからである。(2-2).のニトリルをLAH還元するとアミンが得られるが、単離することなく2,4-dinitrobenzenesulfonyl chlorideでアミンを活性化して (2-1) を得た。 インドール部分 (1-1) とアミン部分 (1-2) のカップリングは光延反応で行い、高収率で目的物 (2-1) が得られた。 (1-1) のBoc基の除去とジヒドロフランの水和はMe2S存在下でTFAを用いて行った。Me2Sの役割はt -ブチルカチオンがインドール環を攻撃しないようにするためである。得られた (1-2) を単離することなく5当量のピロリジンで処理すると2,4-dinitrobenzenesulfonyl基が5分以内にはずれてアミン体 (2-2) が得られた。さらに反応液の温度を60度に昇温して2時間加熱すると、(次ページへ) 環化体 (1-2) が得られた。(1-2) の水酸基の脱水はPPh3-CCl4/MeCNで加熱するという条件を用いたが、再現性(収率)に問題を残すものの何とか目的物 (2-2) を得ることができた。ここでフェノールのメシル基をアルカリで加メタノール分解し、t -BuOK-MeIという、加熱する必要のない条件でフェノールをメチル化して11-methoxytabersonine (2-1) を得た。 (1-1) の酸化はミラノ大学のDanieli教授(奥さんがPradaのファミリー)が報告したとおりbenzeneseleninic anhydride [(PhSeO)2Oを用いてベンゼン中で加熱した。エナミンが反応して、続くセレノキサイド脱離と水の共役付加によって (1-2) が生成する。しかし、問題は次の反応で (1-2) のエナミン部分をMCPBA酸化して (2-2) に変換するのだが、Danieli教授は55%の収率と報告したところ、Vermont大学のKuehne教授はその条件では全く (2-2) が得られなかったと報告した。塩基をK2CO3からNaHCO3に代えてより低温で反応させると20-59%の収率が得られるということだった。私たちがKuehne教授の条件を追試したところ目的物は0-17%の収率で、とてもKuehne先生の収率は得られなかった。様々な条件を検討した結果、溶媒を10%MeOH-CH2Cl2に変更することでコンスタントに60%以上の収率で (2-2) が得られることを見出した。しかし、どうしてこの条件が良いのか、提案した私もよく分からない。(2-2) を還元的メチル化の条件で (2-1) に変換したが、三級アミンがN-オキサイドになっている可能性も考えられたので、NaHSO3処理で念のため還元した。 (1-1) の水酸基をアセチル化して第二世代インドール合成法を用いた(-)-vinblastineの全合成が完了した。第一世代のインドール合成法を使った全合成は後日アップロードする予定だが、いつのことになるかは?? (-)-vindoline (1-2) は150 mgほど全合成で調達できたが、(+)-vinblastine (1-1) の全合成のためにはカップリング相手を合成しなければならない。University of British Columbia (UBC) のJim Kutney教授はピペラジン環を有する (2-1) と (1-2) をカップリングさせると連結部位の立体化学が逆になることを報告している。 Freiburg大学のGottfried Schill教授は(私見では)ビンブラスティンそのものの全合成は目指していなかったと思うが、ピペリジン環を有さないラセミ体のモデル化合物 (1-1)とビンドリンとのカップリングで (1-2) と (1 -3) を1:1の混合物として得たことを報告した。最初この論文を目にしたときに (1-1) がラセミ体であることに気が付かず、立体化学が全然制御されていないと思った。よくよく読み返してみるとラセミ体で1:1になるということは完全に立体化学が制御されていることが分かった。MM2計算で (1-1) の安定配座を解析したところα面が空いていることが分かった。彼らはカップリング後にCbz基を除去してピペリジン環 (2-1, 2-2) を構築している。 (1-1) はベルバナミンというらしいが、前例をもとにしてピペリジン環は後で構築することにして (1-2) を合成することにした。 第二世代インドール合成法を基にした (1-1) の逆合成解析は図のとおり。11員環の構築はノシル基に任せることにすると (1-2) となる。インドール環の構築にはチオアミド (2-2) が必要であり、そのためには (2-1) のようなエステルを合成する必要がある。この光学活性エステルの合成ルートはいくつか私も考えたのだが期待どおりには進まなかった。また別のアイデアを提案したところ、横島君はどうも乗り気ではなく、彼が独自に考えたルートで合成してみたいという「直訴」があった。まあ、博士課程の学生が是非これでやりたいっていうのを止めるのも大人気ないので、それでは自分でやってみろと進めさせたのが次ページからのルートである。 市販のn -butyraldehyde (1-1) にクラシカルなMannich反応を行うとα,β-不飽和アルデヒド (1-2) が簡単に手に入る。Et3Nを少量加えているのはMe2NH・HClが少し酸性に傾いていることがあり、中和して反応性を高めるためだ。Rice大学化学科のAdvanced Organic LabでClaisen-Johnson転位を課題にした時にこの反応を繰り返して大量の不飽和アルデヒドを私自身が作ったことがある。Mannich反応ではMe2NHCH2体が水溶液になっていることが多く、多少不飽和アルデヒドが油として上層に出てくることもあるが、そのまま加熱し、言わば水蒸気蒸留して目的物を取り出す(蒸留された水層にNaClを加えるのを忘れないように)。ジメチルアミンの塩酸塩は反応フラスコに残るので、これにアルデヒドとホルマリンを加えれば次のMannich反応に使えるってわけだ。次に (1-2) をLAH還元してアリルアルコール (1-3) を得た。このアリルアルコールをClaisen-Johnson転位させ、得られたエステルを加水分解して (2-1) が得られた。プロピオン酸を使うと転位条件でエステル化されてしまい反応が途中で止まることがあるが、その場合にはp -nitrophenolを使うことがある。カルボン酸をPivClとの混合酸無水物にしてからEvansの不斉補助基のリチウム塩 (2-2) を加えると (2-3) が得られる。 Evansの条件で (1-1) のアクリロニトリルへの付加を行って (1-2) を得た。次にNaBH4還元でアルコール (2-2) に変換し、シリル基で水酸基を保護して (2-1) に導いた。 ニトリル (1-1) のDIBAL還元は初心者でも確実にアルデヒドを高収率で得られるくらい信頼できる反応である。得られたアルデヒドをH2NOHでオキシム (1-2) に変換した。これをNaOClで処理する常法を使うと非常に反応性の高いニトリルオキサイド (2-2) が生成する。t -butyldiphenylsilyl基 (TBDPS) というデッカい保護基はpseudoequatorial配座をとりやすく、期待どおり1,3-双極子付加反応が進行して目的とするイソオキサゾリン (2-1) のみが得られた。 イソオキサゾリン (1-1) を酢酸中亜鉛還元するとN-O結合が切れ、イミンの加水分解を伴ってケトン体 (1-2) が得られた。これを酢酸中MCPBAでBaeyer-Villiger酸化すると7員環ラクトン (2-2) を得たが、CH2Cl2を溶媒とする通常の条件では反応が起きなかったと横島君は博士論文に記載している。7員環ラクトンは開環しやすく、メタノール中炭酸カリで処理すると容易にメチルエステル (2-1) を与えた。 (1-1) のジオールの保護にアセトナイドを使うと脱保護時にTBDPS基も部分的に外れてしまうので、便宜上TESClで1級アルコールを保護し、なかなか反応しない3級アルコールを同一フラスコ中にTMSClを加えることでTMS化して (1-2) を得た。1級アルコールのTMSエーテルは簡単に加水分解されてしまうが、3級アルコールの場合はかなり安定で少々の酸性条件でも持ちこたえる。メチルエステル (1-2) をLDAで脱プロトン化してイソチオシアネート (1-3) を加えるとチオアミド (2-1) が得られた。これをラジカル反応の条件に付すとインドール (2-2) に変換することができた(第二世代インドール合成法)。 (1-1) のインドールNHはBoc2O-DMAPでBoc化しておかなければならない。これは後に光延反応を使ってノシルアミドと反応させる時にシクロプロパン化が優先してしまうのを防ぐためだ。なお、インドールNHを保護するのはこのBoc化が最適で、例えばスルホンアミドにしようとすればNaHでアニオンを作らなければならない。次に含水酢酸で加熱することでTHP基とシリルエーテルを同時に脱保護してトリオール (1-2) を得た。このトリオール (1-2) の1,2-ジオール側の1級水酸基をトシル化するのはMike MartinelliのTsCl-Bu2SnO-Et3Nという条件を使った。Mikeは岸研でポスドクをやってEli Lillyに長く勤めた私の友人でなかなか良い男である。Bu2SnOを使って1,2-ジオールの立体的により空いているアルコールをエーテル化する方法は以前から知られていたが、これをトシル化に応用したものだ。環状スズエステル構築してアルコールを活性化する反応機構である。得られたモノトシレート (1-2) をアジドに変換しようとNaN3と加熱したところ、エポキサイド (2-2) が得られただけなのでこれを使うことにした。反応条件としてはDMF中でNaHCO3存在下に加熱して (2-2) を得た。 ダイマーの生成を防ぐために2当量のNsNH2を用いてアルコール (1-1) をアミン誘導体 (1-2) に変換した。出典は定かでないが光延反応はベンゼン(又はトルエン)、CH2Cl2、THFの順に反応速度が落ちてくると記憶している。(1-2) をK2CO3存在下DMF中90度で終夜加熱することで11員環中間体 (2-1) が高収率で得られた。次にBoc基とTBDPS基を酸性条件(TFA)で外して (2-2) を得た。 3級アルコール存在下で1級アルコールをトシル化するのは関学大の田辺陽さんの条件を用いた。Me2N(CH2)3NMe2トシル化の加速効果がどこに起因するのかイマイチ分からないが通常のEt3NやPyを用いる場合よりは好結果を与えた。3級アルコールを保護しておかないとビンドリンとのカップリングが進行しないので、ここでは簡単に除去できるトリフロロアセテート (1-2) に変換しておいた。(1-2) のCH2Cl2溶液に0度で1.1当量のt -BuOClを加え、10分後にシリカゲルのカラムを通してから溶媒を留去した。得られた (2-2) と0.9当量の我々が合成した(-)-vindolineのCH2Cl2溶液に、0度で10当量のTFAを加え10分後に室温に昇温して50分後に後処理、カラム精製したところ(-)-vindolineベースで97%の収率でカップリング生成物 (2-1) が得られた。 (1-1) のトリフロロアセテートを加メタノール分解し、さらにHSCH2CH2OH-DBUでノシル基を外して (1-2) を得た。最後のピペリジン環構築はベルバナミン部位のインドール環の3位が関与して副生物が生成した。横島君はこの副生物生成を抑制するためにかなり苦労したがH2Oがピペリジン環生成を加速することに気がついて結局i -PrOH-H2O中で環化を行うことで最適の収率が得られることを見出した。完全全合成された(+)-vinblastineの旋光度は文献値と良い一致を見せた。助教授だった徳山さんをはじめ学生諸君がしっかりと働いてくれたおかげで完成できた一大プロジェクトだった。