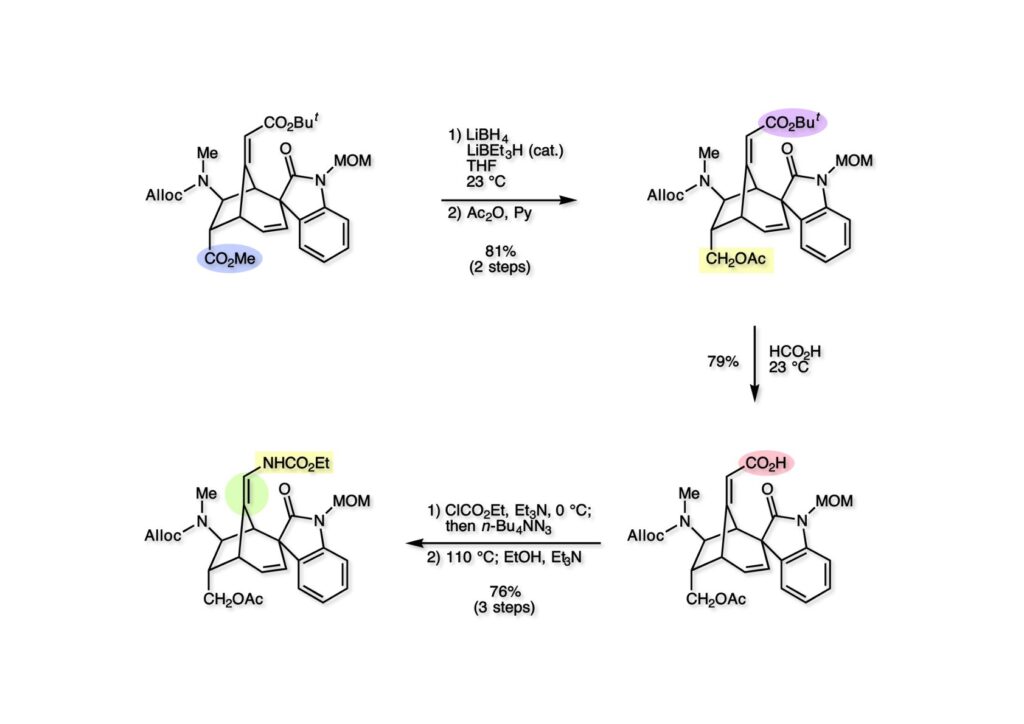
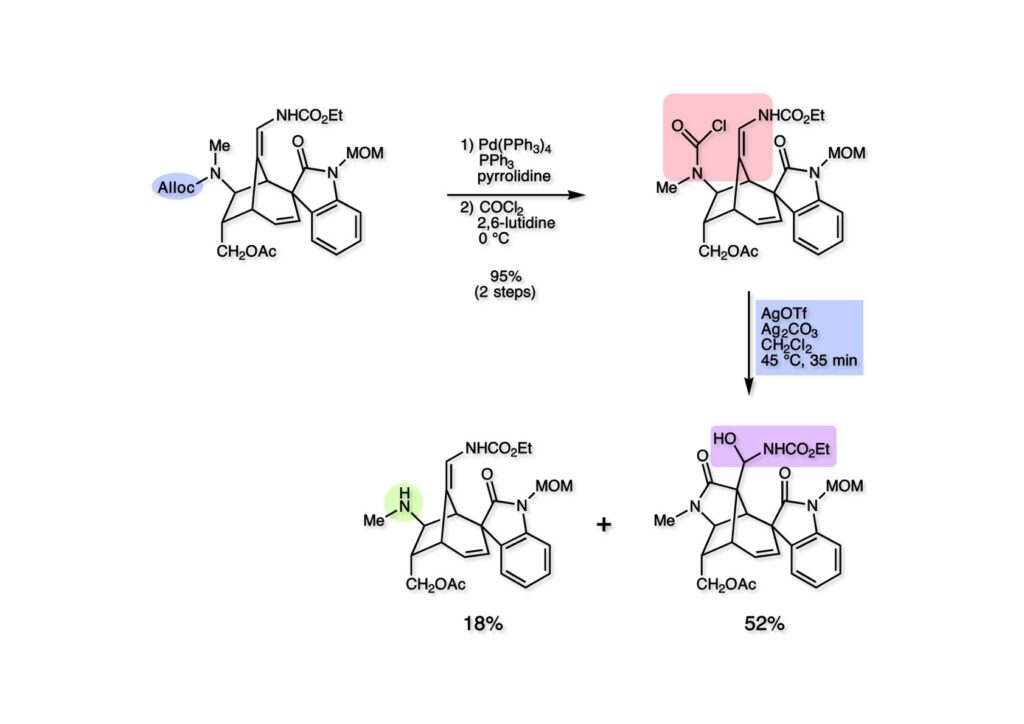
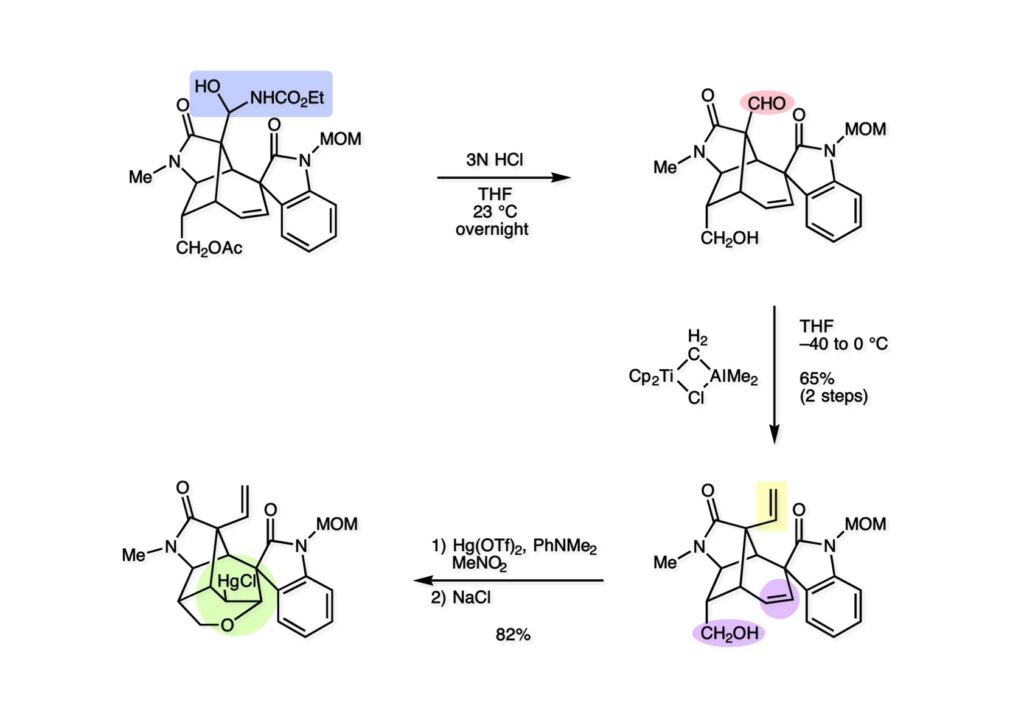
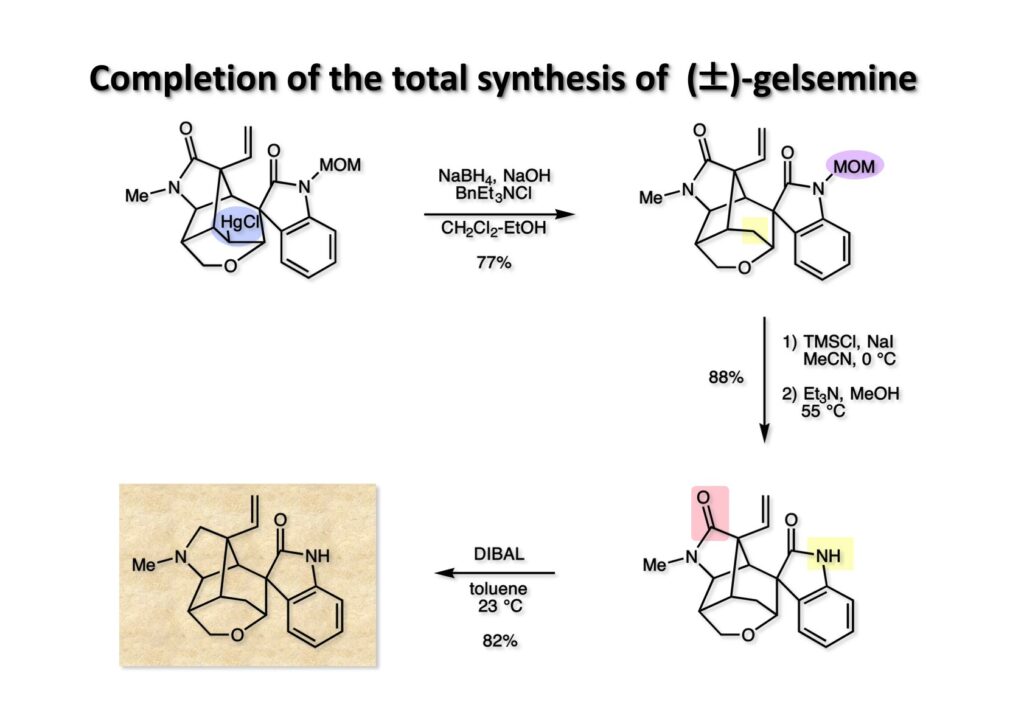
ゲルセミンは私が学生の頃から知っている有名なアルカロイドで、生物活性は大したことはなくとも構造的に非常に面白い化合物という印象だった。これを全合成してみようという気は毛頭無かったが、1990年頃にNIHの研究費審査員(4年任期)をやっていた時に某教授のプロポーザルの担当になった。その教授はすでに6、7年は全合成に挑戦しており、プロポーザルを読むとまだ誰も全合成に成功していないことが分かった。「そろそろ真打の出番かな?」なーんて思って自分なりに合成戦略を考えてみた。勿論、その教授のプロポーザルに書いてあるアイデアを盗用しようなどとは微塵も思っていないし、実際、似ても似つかない合成となったが、動機が不純だ!と言われれば、多少、そうかな、と。このプロジェクトはGang Liuという北京大学出身の院生で、当時としてはシティーボーイという中国人らしく見えないスマートな若者で(勿論現代の中国の若者は昔とは全然違った感じなのは言うまでも無いが)、手際よく全合成を完成させた。日本に帰って来てから天然物討論会で初めて口頭発表したのは記憶に残っている。その後、光学活性のゲルセミンを合成してみようと思い、東大で最初に研究室に入った学部生の横島聡君(現名大創薬科学研究科教授)に託したところ見事に仕上げてくれた。
“Stereocontrolled Total Synthesis of (±)-Gelsemine,” T. Fukuyama and G. Liu, J. Am. Chem. Soc. , 118 , 7426 (1996).
“Enantioselective Total Synthesis of (+)-Gelsemine: Determination of Its Absolute Configuration,” S. Yokoshima, H. Tokuyama, and T. Fukuyama, Angew. Chem., Int. Ed. , 39 , 4073 (2000).
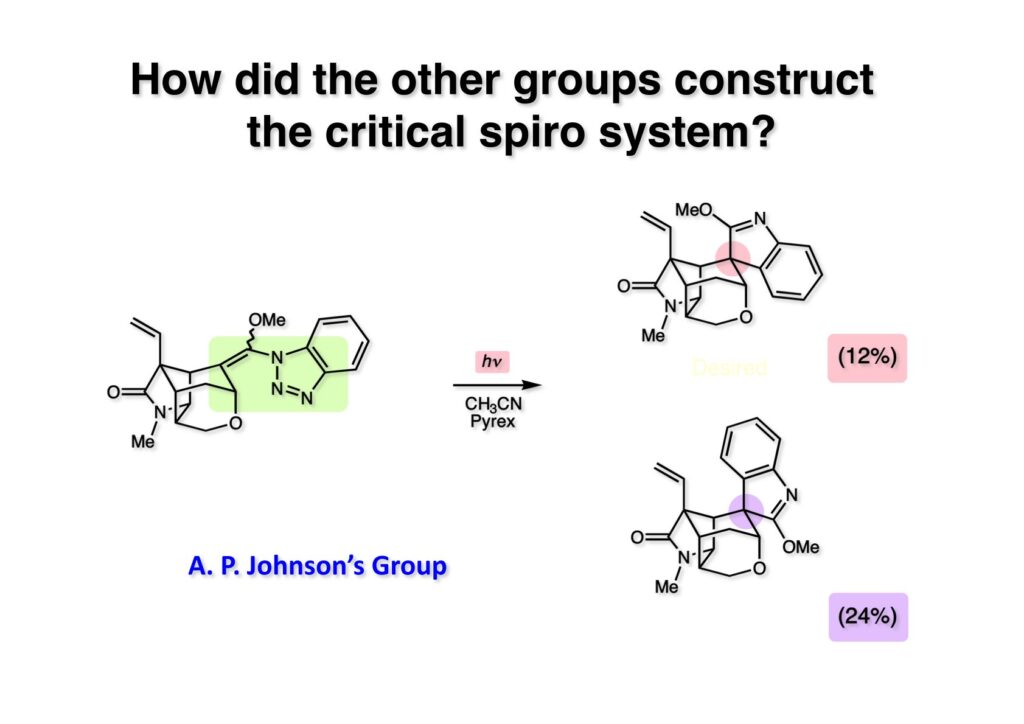
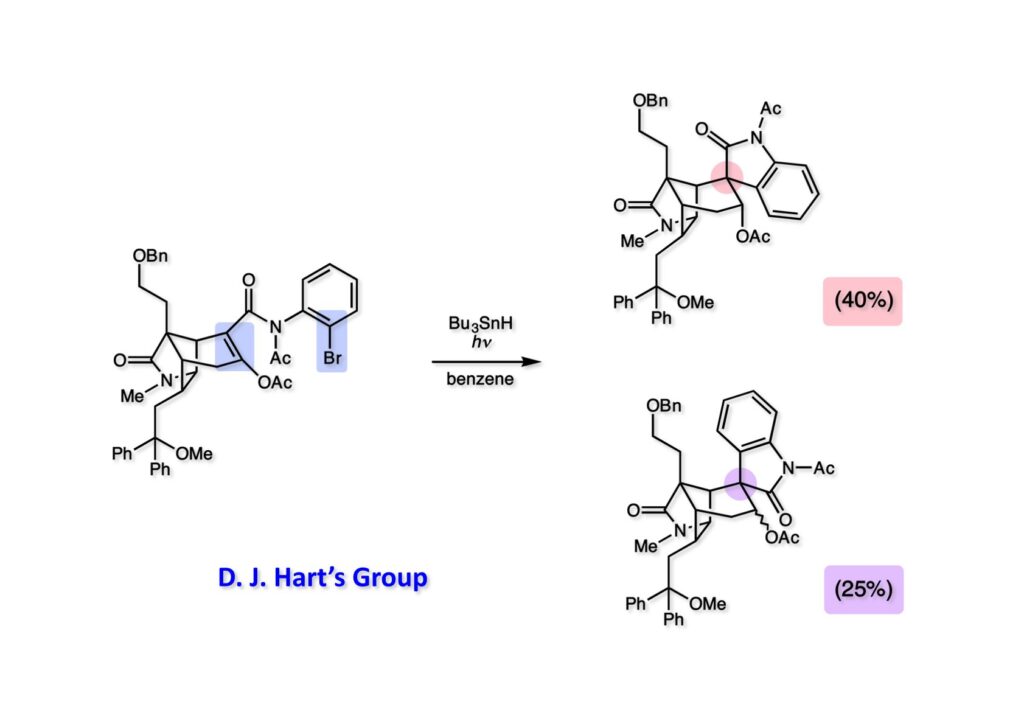
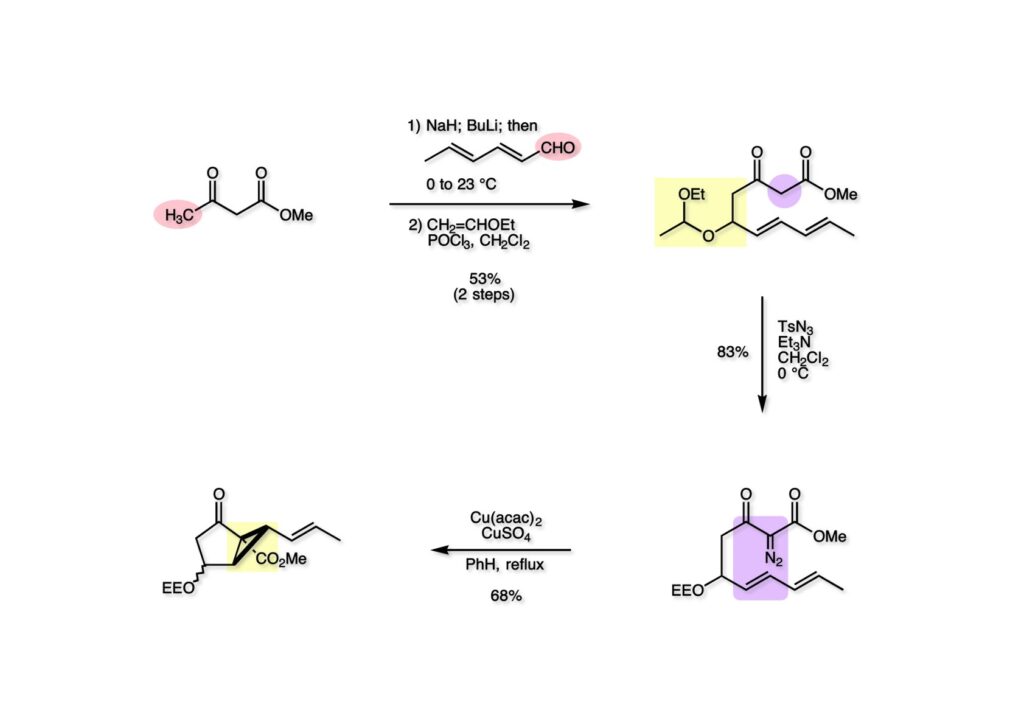
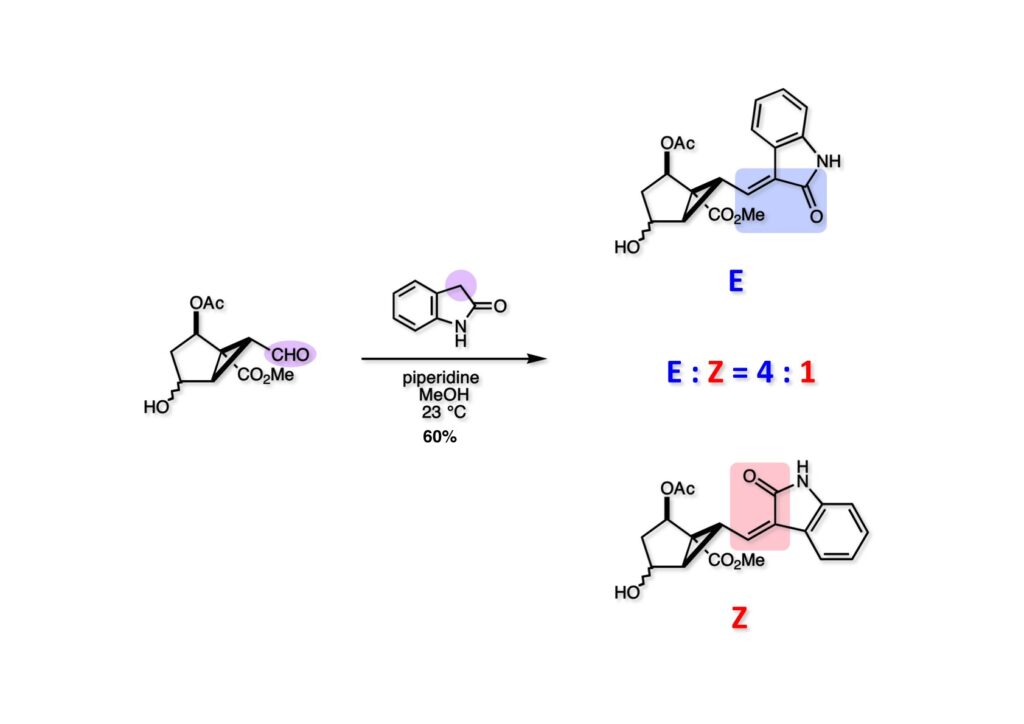
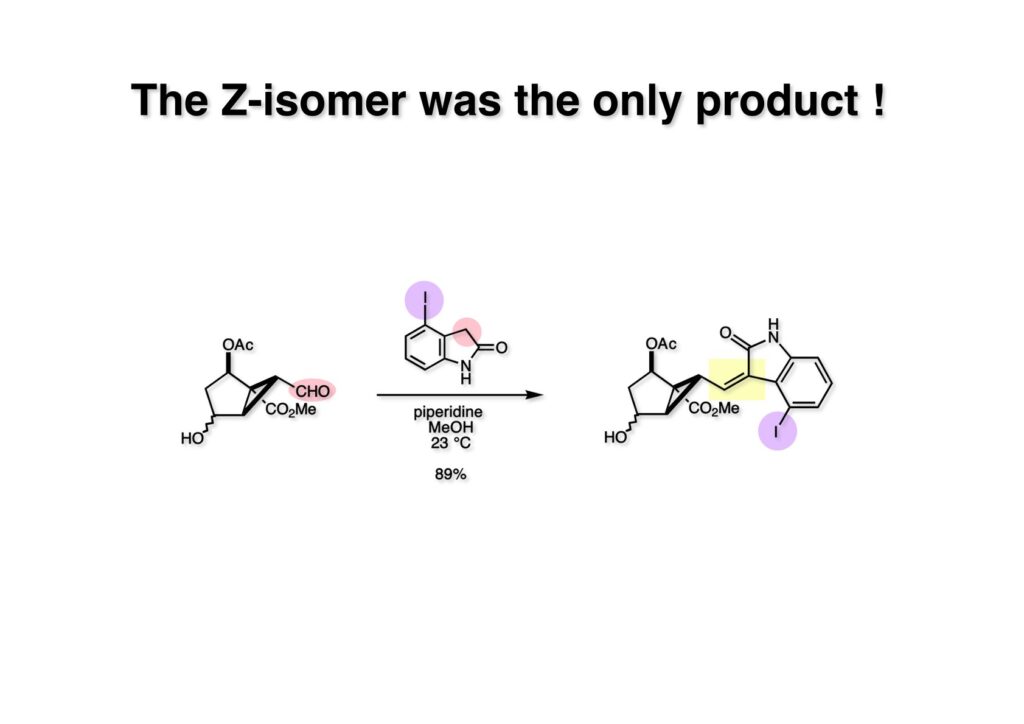
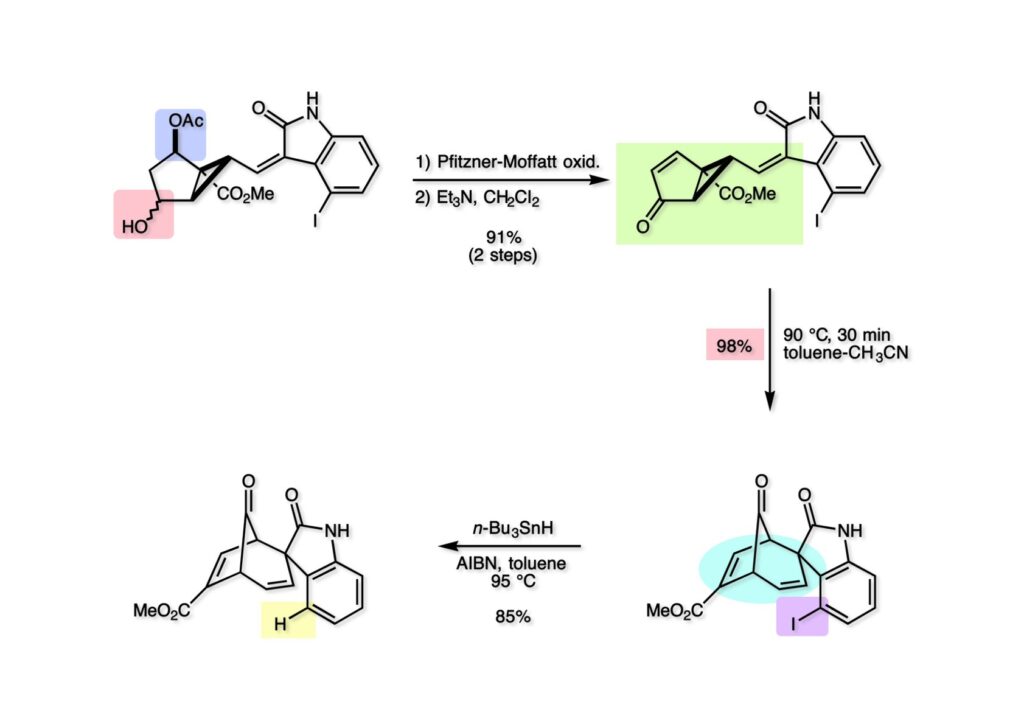
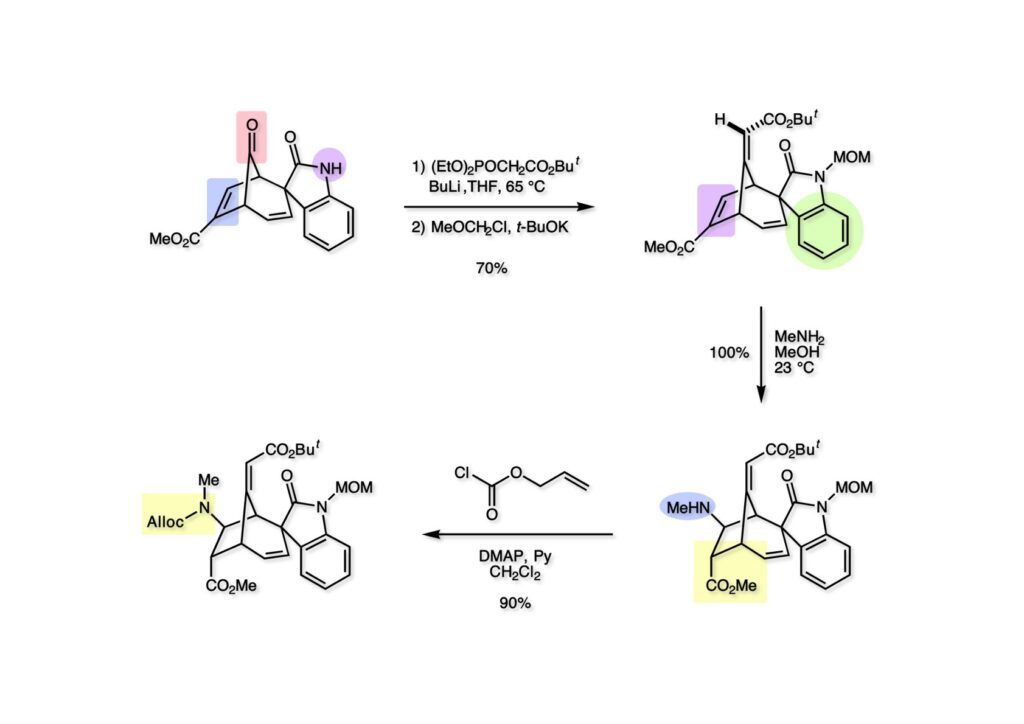
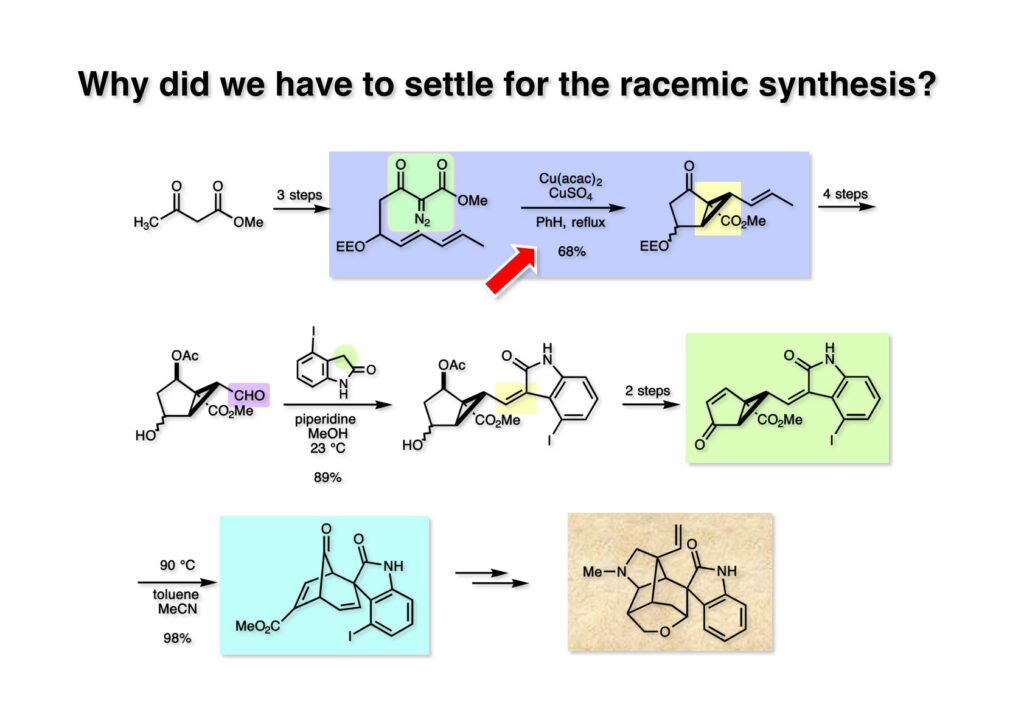
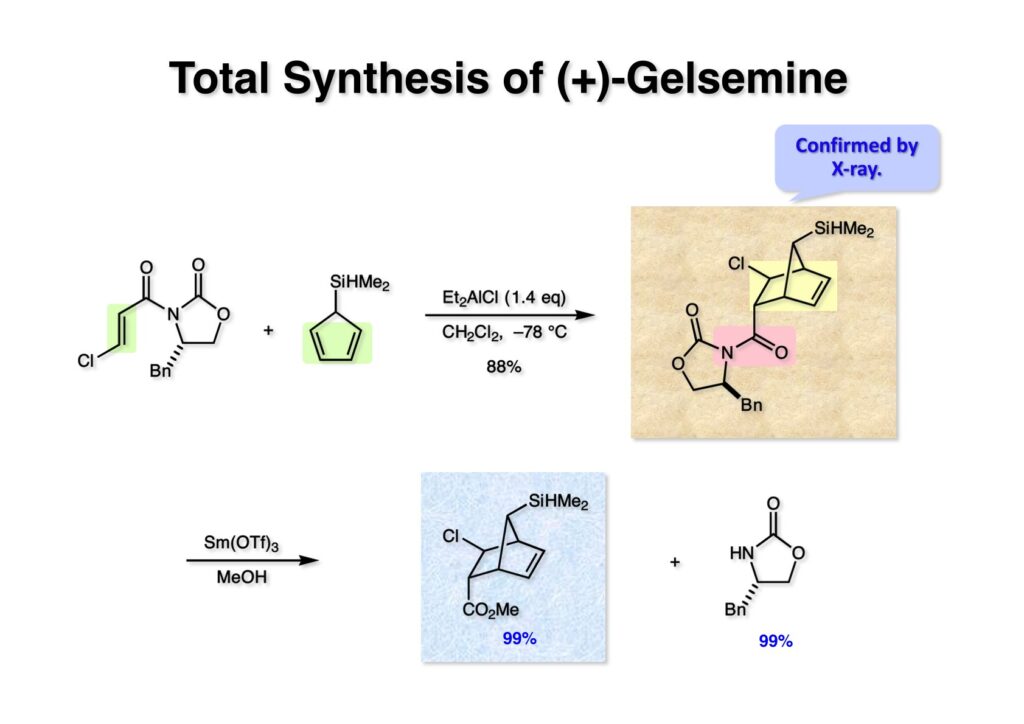
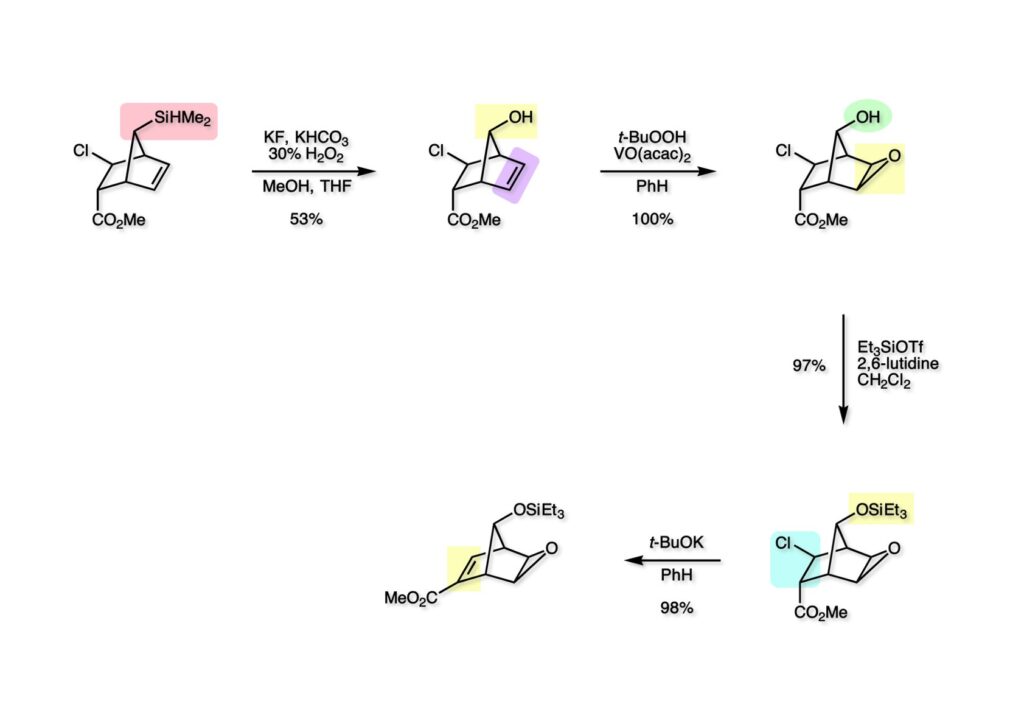
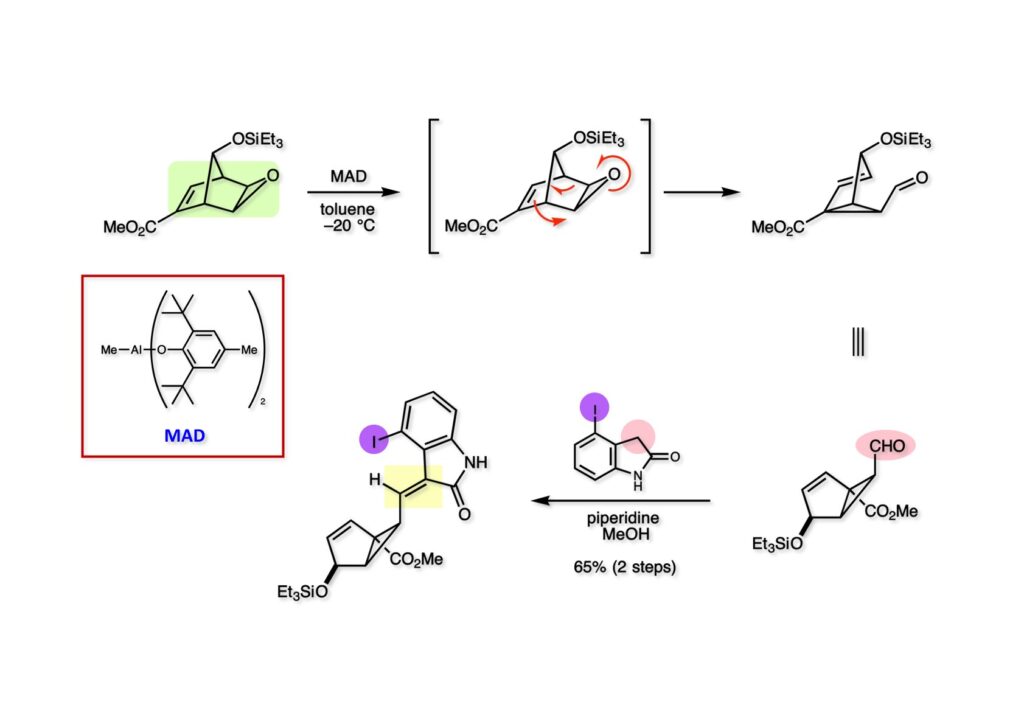
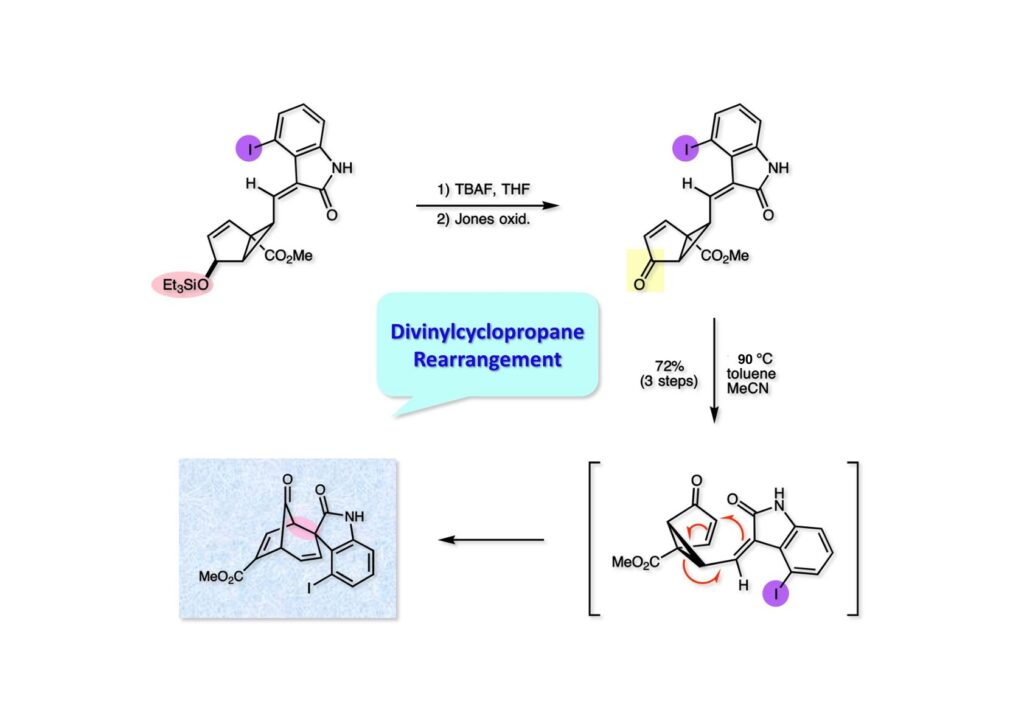
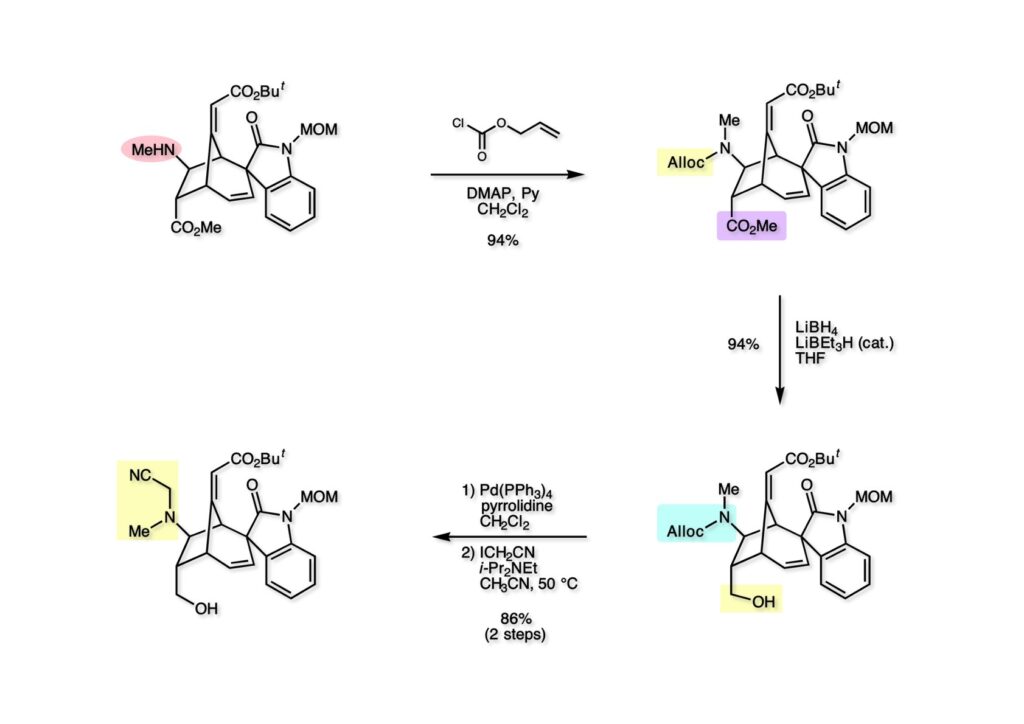
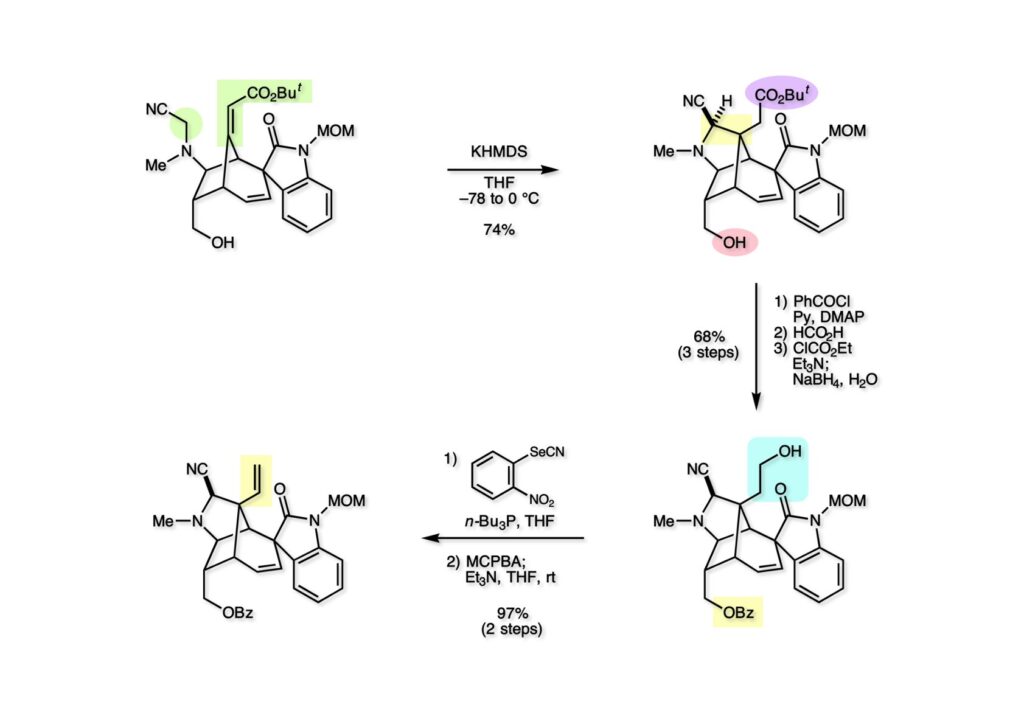
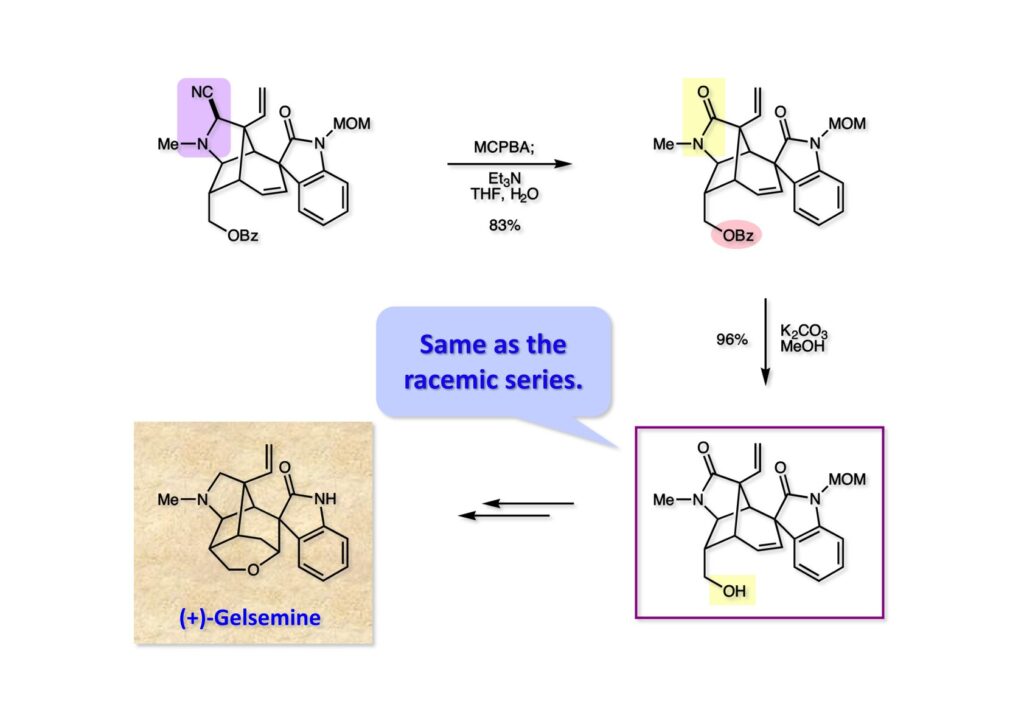
我々が全合成研究を開始したのは1993年の前半で、翌年には3つのグループが次々に全合成を報告した。Peter Johnson (University of Leeds)、Nico Speckamp (University of Amsterdam)、そしてDavid Hart (Ohio State University)のグループである。いずれも10年近く全合成をやっていて執念でゲルセミンに到達した。先行する全合成ではスピロ環の立体制御に問題があり、まだまだ改良が必要だという印象を受けた。 Peter Johnsonにリーズ大学で会った時にTFが岸研の院生だった時にWoodward研でポスドクをやっていたそうだ。全合成も少しはやっていたが、研究の中心は酵素のポケットに入るような小分子をコンピュータでデザインするプログラム開発をやっていて、いくつかの製薬会社が彼のプログラムを買っていたそうだからお金持ちになったかな? SpeckampはHeck反応を使って (2-1) からスピロ環を構築したが目的物 (1-1) は60%、いらない化合物 (3-1) は30%の収率だった。彼らは (2-1) に辿り着くまでにかなりの工程数を要したが、Heck反応の条件はOvermanがゲルセミンの合成研究として報告したのを使って成功した。中間報告を出しすぎると手の内が知られてしまうのは仕方ないことだろう。 David Hartは昔からの友達でNIHの研究費審査員でも重なっていて、ゲルセミンの全合成がなかなか終わらないのでフラストレーションが溜まっているという愚痴を聞かされていたものだ。彼は早くからラジカル反応を応用した天然物の合成をやっていて、ゲルセミンの全合成研究もその一環だった。Speckampと異なり (2-1) をラジカル反応でスピロ環を構築して目的物 (1-1) を40%、要らない方 (3-1) を25%の収率で得ている。但し、Davidは (3-1) のアセテートをアルカリ加水分解してretro-aldol-aldol反応で目的とするスピロ環に異性化することに成功している。後にOvermanはこの異性化反応を使って全合成を終えた。 最初の1年は2つのルートを試みたがうまく行かなかった。その頃見つけた第一世代のインドール合成法を使おうと思い、bicyclo[3.2.1]中間体を構築しようとした。ところがアセチル基とアルデヒドを分子内アルドール反応で7員環を作ろうと思ったが、最初にアルデヒドが反転し、次にアセチル基も反転するという間抜けなことが起きて諦めてしまった。「あーあ、(2-3) のような中間体が欲しいなー」とGangの机の横の黒板に構造式を書いておいた。ある朝、Gangの居る実験室に入ってその構造式を見た時に、あっと思った。これって3つの矢印を使えば (2-2) に変換することができる、ということはdivinylcyclopropane rearrangementを用いれば (2-2) から (2-3) へ導くことができるということだ。(2-3) が手に入ればメチルアミンを共役付加させて (1-3) にし、うまくピロリジン環 (1-2) を構築し、そして6員環エーテルを作ればゲルセミン (1-1) が合成できるという展望が開ける。 前ページの (2-1) の合成にはヒントとなる報告があった。相模中央研究所の近藤聖博士がプロスタグランジンの合成に用いたルートだ。市販のmethyl acetoacetate (1-1) をNaHでナトリウム塩にして、さらにn -BuLiを加えるとジアニオンが生成する。それに不飽和アルデヒド (1-2) を加えて室温まで昇温すると末端で付加反応が起き、さらに水酸基を保護して (1-3) が得られた。ここで今でもモヤモヤしているのは、アルデヒドへの付加反応を-78度で行ってクエンチすると反応が起こらなかったことだ。そんな筈はないとGangに言ったんだけど。(1-3) をTsN3-Et3Nでジアゾトランスファーさせるとジアゾ化合物 (2-2) が得られた。次に銅触媒を用いてシクロポロパン化することで (2-1) を得た。 ケトン (1-1) をNaBH4で還元してアルコール (1-2) とし、無水酢酸でアセテートとして保護してからethoxyethyl (EE) 基を酸で脱保護して (2-2) を得た。ここでオレフィン (2-2) をオゾン分解してアルデヒド (2-1) に変換した。 アルデヒド (2-1) と市販のオキシインドール (oxindole, 2-2) をピペリジン存在下でKnoevenagel縮合したところE 体 (1-1) とZ 体 (3-1) の生成物が4:1の比で得られた。試しに主生成物.(E 体)を後述するルートでdivinylcyclopropane転位に付したところ目的物でないスピロ体が生成することが判明し、Z 体を使わなければならなくなった。 分子模型を組んでみるとオキシインドールの4位に大きな基が存在すれば、それを避けるようにZ 体が主生成物になるだろうと予測できた。最初は4-bromo-oxindoleを出発物にしたが、ラジカル反応による脱ブロモ化がかなり困難であったので、4-iodo-oxindole (1-2) を使うことにした。予想通り、アルデヒド (1-1) と (1-2) とのKnoevenagel縮合は目的とするZ 体 (1-3) のみを与えた。 (1-1) の水酸基の酸化はPfitzner-Moffatt酸化 (DMSO-DCC-TFA-Py) を採用し、得られたケトンにEt3Nを加えると酢酸が脱離して不飽和ケトン (1-2) が得られた。これをトルエンーアセトニトリル混合溶媒中で 90度に加熱するとほぼ定量的に目的物 (2-2) に変換することができた。アセトニトリルは出発物の溶解性を高めるために使用した。この転位反応は本全合成の最重要段階で先行3全合成では制御できなかったスピロ環の立体化学の問題を解決した。用済みになったヨウ素原子はラジカル反応で除去して (2-1) を得た。 このbicyclo[3.2.1]体 (1-1) の8位のケトンのままだとMeNH2やEtSHが不飽和エステルにMichael付加したが、NaBH4で還元した化合物については全く共役付加が起きなかった。これは立体障害と環の歪みの減少に起因するかもしれない。そこでHorner-Emmons反応でケトン (1-1) から不飽和エステルに増炭し、同じポットでMOM基を導入して (1-2) を得た。最初はKHを使ってアニオンを出したところ4:1の混合物になったが、BuLiを用いたところ単一の生成物となった。(1-2) の立体化学はNOEで決定した。この図ではそれほど明らかでないが、(1-2) のインドリノンのベンゼン環はビシクロ体の下面を大きく覆っており共役付加は上からしか起きない。見込み通り、メタノール中でメチルアミンを加えると定量的に目的物 (2-2) を与えた。(2-2) のMOM基の代わりにMe基を持つ化合物を使ってアミンを足場にしてラジカル的とカルバニオン的に5員環を構築しようと努力したが環化は起きなかった。ラジカル反応は可逆的なので、おそらく環化した中間体が歪みによってエネルギー的に不利だったのかもしれない。エステルのカルバニオンも共役付加しなかった。ところが第二合成において、横島君はニトリルのカルバニオンの共役付加に成功したので、Gangの努力が足りなかったのかもしれない。取り敢えず、ここではアミン (2-2) をAlloc基で保護して (2-1) を得た。 とにかく不飽和エステルを使っての5員環構築は諦めて、「押しても駄目なら引いてみよ」ということで電子豊富なオレフィンに変換することにした。(1-1) のCO2Me基は塩基性で簡単にエピめってβ配置になってしまうのでLiBEt3H-LiBH4で還元してアルコールにし、さらにアセチル化して (1-2) に変換した。次に (1-2) をギ酸処理によってt -ブチルエステルをカルボン酸 (2-2) に導き、さらに混合酸無水物経由で酸アジドにした。混合酸無水物が加水分解されないようにCH2Cl2に可溶なn -Bu4NN3を用いた。n -Bu4NXの製法はn -Bu4NHSO4とNaXをエタノール中で混合し析出したNa2SO4を濾過してエタノールを留去すれば出来上がり。結構便利な試薬となり得るので覚えておくと良い。得られた酸アジドをトルエン中で加熱し、得られたイソシアネートをEt3Nとエタノールで処理してウレタン (2-1) を得た。 (1-1) のAlloc基を常法で除去し、得られたアミンをホスゲンでカルバモイルクロライド (1-2) に変換した。日本ではホスゲンのボンベは実験室レベルでは入手できないが、アメリカでは小さいボンベで容易に購入できた(今は知らない)。ここで驚いたのは (1-2) の安定性で、シリカゲルTLCで精製しても全く分解しなかった。通常の酸クロライドでは考えられないほどの安定さだ。ということは反応性が低いということだが、とにかく銀塩で無理やりアシリウムイオンを生成させて電子豊富なエナミド系オレフィンに噛みついて欲しいという計画だった。反応条件としては無水のCH2Cl2を溶媒にしてAgOTfとAg2CO3を用いて酸性にならないように考慮した。加水分解されて (2-1) に戻ってしまったのが18%、C-C結合形成で5員環構造を有する化合物 (2-2) が52%の収率で得られた。アルデヒドが得られると思っていたが、まるでアルデヒドのピークは見られず、Et基のシグナルが見えたのには驚いた。(2-2) の意外な安定性はOHとNHCO2Etのいずれもがカルボニル基への分子内水素結合をしているからと考えられる。 (1-1) はTHF中3N塩酸で終夜処理することでアルデヒド (1-2) に変換することが出来た。このアルデヒドからビニル基に変換する反応はWittig反応で簡単に行くだろうと思ったがPh3PCH2では全く反応しなかった。おそらく立体障害によるものと思われる。そこでTebbe試薬を調製して反応させたところ無事に目的とするビニル体 (2-2) が得られた。(2-2) のオレフィンとアルコールからのエーテル環形成はオキシ水銀化反応を用いた。分子モデルを見てみると5員環形成の方が有利に思えたがオキシ水銀化は可逆反応なのでおそらく熱力学的に安定な6員環エーテルに収束したのだろう。このオキシ水銀化には通常のHg (OAc)2は役に立たず、西沢麦夫さんの開発した強力なHg(OTf)2でうまく進行した。これにNaCl溶液を加えると安定な (2-1) が得られた。いつも不思議に思うのだがオキシ水銀化反応をTLCで追跡すると、出発物の消失は分かるが生成物がまともなスポットで観察されたことがない。これにNaClを加えると綺麗なスポットが現れる。何でだろう?? さて、(1-1) の脱水銀化は予想外に手間取った。通常の条件でNaBH4還元すると環化前のアルコールに戻るのが主反応で、目的物 (1-2) は少ししか得られなかった。困ったGangは独断でアムステルダム大学のNico Speckampに脱水銀化の秘訣を尋ねてしまった。今みたいに実験条件がSupporting Informationが掲載されてない頃の話だが、私自身は簡単にギヴアップしない主義なのでちょっと忸怩たる思いがある。Gangが得た情報はphase-transfer触媒を使うものでSynthesis, 891 (1979) にCNRS(フランス)のケミストが報告していた。確かにこの条件を用いると目的物 (1-2) が主生成物として得られてきた。次は (1-2) のMOM基を除去する必要があったが2段階で実行した。まずNaI-TMSClで反応容器内でTMSIを作り、それがMOM基を攻撃してMeO基の脱離によりICH2N体が生成すると思われるが後処理によりHOCH2N体が得られる。この化合物は安定でホルムアルデヒドを追い出すにはメタノール中Et3Nを加えて加熱する必要があった。得られた (2-2) は21-oxogelsemineでこれも天然物であり、スペクトルは報告されたデータと一致した。この化合物からゲルセミン (2-1) への変換は既知反応で、DIBAL還元することにより容易にゲルセミンが得られた。 第一世代のゲルセミン全合成はラセミ体の合成となったが、その理由は分子内シクロプロパン化 [(1-2) -> (1-3)] の不斉化が出来なかったからだ。今だったら不斉触媒を使って (1-3) の光学活性体が得られるかもしれないが、文献調査はしていない。1995年に帰国してからも光学活性ゲルセミンの合成には興味を持っていたので別のルートを考えてみることにした。この合成は腕の悪い学生にはとても頼めないレベルで、最初の学部生で何事もキッチリやる横島君に頼んだ。 光学活性ゲルセミンの全合成ルートを考えるにあたり、以前ライス大学の院生にcumulative examinationの反応機構問題としてMeinwald転位を出題したことを思い出した。これはnorbornadieneをモノエポキシ化した化合物を酸触媒によって転位させるとbicyclo[3.1.0]hex-2-ene-6-carboxaldehydeが生成するという問題で、紙に書いてみるとまさしくゲルセミン合成の中間体になりうる構造だった。この反応を元にして不斉合成のスキームを考えた。Evansの不斉補助基を用いたdienophile類はシクロペンタジエンとのDiels-Alder反応で良好な成績で付加体を与えることが知られていた。そこで (1-1) と5-dimethylhydrosilyl-1,3-cyclopentadiene (1-2) のDiels-Alder反応を低温下ルイス酸 (Et2AlCl) を用いて行ったところ高収率で単一の付加体 (1-3) が得られた。(1-3) の絶対配置はX線解析によって決定した。(1-2) は東北大の平間さんが開発した化合物だが全合成の論文に引用し損なって失礼した。付加体 (1-3) をメタノール中でSm(OTf)3を使って室温放置したところメチルエステル (2-1) とEvansの不斉補助基 (2-2) がほぼ定量的に得られた。 (1-1).のdimethylhydrosilyl基を玉尾-Fleming酸化してアルコール (1-2) に変換し、Sharpless酸化でオレフィンをエポキシ化すると (1-3) が得られた。ここで水酸基をシリル化して (2-2) を得た。(2-2) をベンゼン中でt -BuOKで処理すると容易に脱HClが進行して不飽和エステル (2-1) が得られた。 ここでMeinwald転位を実行する段階なのだがシリル基で保護していないとブリッジが開裂してフタル酸モノアルデヒドが生成してしまった。(1-1) に種々のルイス酸を作用させたが目的物 (1-3) は得られるものの、そこからさらに転位反応が進行した副生物が生成してしまい収率が低下した。最終的には山本尚酸が開発したMADを用いることでまずまずの収率で転位反応を行うことができた。ここで転位反応がカチオン性のルートを辿るとすればエステルの結合した炭素にカチオンを作るのはエネルギー的に不利なので目的とする化合物が主生成物になることは予想できた。実際は (1-3=2-3) のみが得られた。(2-3) を4-iodooxindole (2-2) とKnoevenagel縮合して (2-1) を得ることに成功した。 (1-1) のEt3Si基をTBAFで除去し、得られたアルコールをJones酸化することでエノン (1-2) が得られた。第一世代のゲルセミン合成においてはtrans -divinylcyclopropane転位を行ったが、今回はcis -divinylcyclopropane転位となる。いずれの場合も3,3-sigmatropic rearrangementが進行することは知られているが、一般的にcis体の方がより低い温度で転位が起きる。その昔Yale大学のJerome Berson教授の講演を聞いたことがあるが、先生は3,3-sigmatropic rearrangementはdiradical機構で説明されていた。ところが (1-2) は室温で不安定で放置しておくとtrans 体との混合物になってしまった。そこで単離することなくtoluene-MeCN中90度1時間加熱することで単一の目的物 (2-1) を83%の収率で与えた。ここで第一世代の中間体とデータを比較して同一物であることを確認した。 脱ヨウ素化の段階が第一世代とは少し違うが、あとはほぼ同じルートで (1-1) から (2-1) を合成した。 ここも同様に (1-1) から (1-2) を経て (2-2) を合成し、Alloc基の除去後にICH2CNと加熱してシアノメチル体 (2-1) を得た。Gang Liuは主にラジカル反応を用いて5員環を構築することに注力したが環化は全然起きなかった。アミンをアセチル化してLDAを用いて6員環を構築する試みも失敗している。横島君はシアノメチル基にアニオンを発生させて5員環を構築することにした。このアイデアは私が指示した記憶が無いので、横島君が自分で考えたのだと思う。本当は全部私が考えたルートだと言いたいところだが、学生に出し抜かれることもあるというのは、ボスとしては悔しい気持ちと弟子が育っているという喜びとが入り混じった複雑な心境ではある。 シアノメチル体 (1-1) からKHMDAによってカルバニオンを生成させると首尾よく不飽和エステルに共役付加して5員環化合物 (1-2) が得られた。第一世代全合成で忸怩たる思いだったのはブリッジ上にビニル基に変換可能な2炭素が用意されているのにそれを利用できなかったことである。まず (1-2) の水酸基をベンゾエートとして保護してからギ酸処理でt -ブチルエステルをカルボン酸に変換、そして混合酸無水物経由でNaBH4還元することでアルコール (2-2) を得た。次にGrieco-Nishizawa法でアルコールをo -nitrophenylselenideに変換し、MCPBA酸化と続くセレノキサイド脱離でビニル基 (2-1) を構築した。この反応でMCPBAを少し過剰に使ったため、後にラクタムがわずかに生成していることを横島君は見逃さなかった。彼はアミノニトリルをラクタムに変換する反応を発見し、後日論文をパブリッシュすることが出来た。いずれ当HPで反応開発についても解説するつもりであるが、私が全然寄与していない研究を収納すべきかどうか。まあ100%横島君にクレジットすることで許してもらおう。 アミノニトリル (1-1) をMCPBA酸化でアミンオキサイドに変換し、次にEt3Nを加えて室温で4時間反応させることで収率よくラクタム (1-2) を得ることができる。(1-2) のベンゾエートを加メタノール分解して得られたアルコール (2-2) は第一世代に合成したものと一致し、すでに確立したルートで光学活性ゲルセミン (2-1) の全合成が完了した。ゲルセミンの絶対配置は生合成経路から (2-1) のように提示されていたが、合成によってそれが証明された。因みにアミノニトリル (1-1) をNaBH4還元するとゲルセミンと同じメチレン体になるが、残念ながらオキシ水銀化がうまくいかなかったのでラクタムに変換する必要があった。