Hapalindole類もtantazole類同様ハワイ大学のDick Mooreが単離構造決定した海洋天然物で、ちょっと面白い構造を有しているので全合成をやってみようと思った。大した生物活性は無かったと思う。見た目が全てっていう感じでね。Tantazole Bの全合成を達成したLianhong Xuのボーイフレンド(当時は知らなかった)で現在は夫婦でカリフォルニアに暮らしているXiaoqi Chenに全合成をやってもらった。まだインドール合成を開発する前の話で、たまたまインドール骨格が入っているが、特にそれに拘ったわけではない。
“Stereocontrolled Synthesis of (–)-Hapalindole G,” T. Fukuyama and X. Chen, J. Am. Chem. Soc., 116, 3125 (1994).
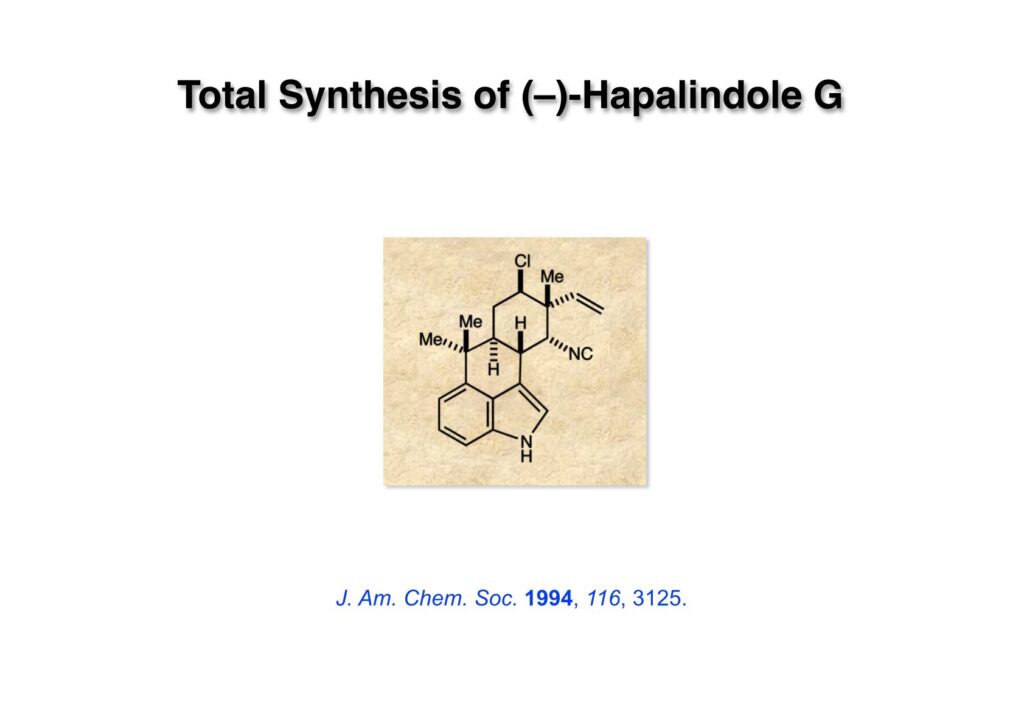
 全合成を開始した時点ではhapalindole J, Q, Uの全合成が報告されていた。しかしClが入ったものは手付かずで、四級炭素の隣にClを如何にして導入するかが私の興味の中心だった。
全合成を開始した時点ではhapalindole J, Q, Uの全合成が報告されていた。しかしClが入ったものは手付かずで、四級炭素の隣にClを如何にして導入するかが私の興味の中心だった。
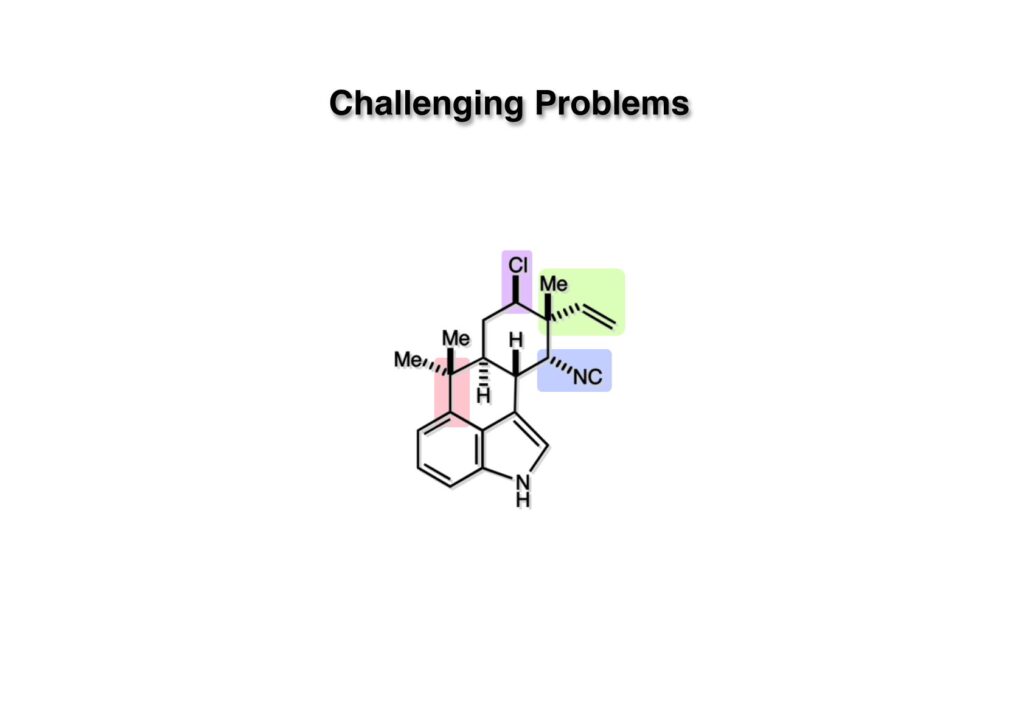 Hapalindole Gの構造上の特徴は前述の(1)四級炭素の隣にClが存在すること、(2)海洋天然物に時々見かけられるイソニトリルの存在、(3)四級炭素に結合したビニル基の存在、(4)インドールの4位に置換基があること、などが挙げられる。
Hapalindole Gの構造上の特徴は前述の(1)四級炭素の隣にClが存在すること、(2)海洋天然物に時々見かけられるイソニトリルの存在、(3)四級炭素に結合したビニル基の存在、(4)インドールの4位に置換基があること、などが挙げられる。
 四級炭素の隣にCl基を導入する方法については一案があった。例えばシクロヘキサンに脱離基が存在する場合、求核置換反応を行うとSN2反応の生成物とともにE2反応でオレフィンが副生物となることが散見される。脱離基はaxialまたはpseudo-axial配座をとってSN2反応が進行するが、隣接する炭素にtrans-Hが存在すればC-H結合とC-脱離基結合のσ*軌道の重なりが大きくなりE2反応が起こりやすくなる。一方、シクロヘキセンオキシド(エポキサイド)のSN2置換反応においては、このような軌道の重なりが小さいためにオレフィンの生成は抑制気味となる(全く生成しないわけではない)。とにかく、この事実は頭の中に入っていたので、シクロプロパン環をCl-で開裂させることを企画した。まず、市販の(+)-carveol (1-1) をマロン酸エステルに変換してジアゾトランスファーにより (1-2) を得た。これを銅触媒 (2-1) 存在下で加熱してシクロプロパン誘導体 (3-2) を得た。(3-2) の環開裂は少し過激な条件を必要としたが、DMF中でLiClとカンファースルホン酸 (CSA) 存在下で140度に加熱することで目的物をまずまずの収率で得ることができた。脱C O2Me基はKrapchoの条件に近いから当然のことである。
四級炭素の隣にCl基を導入する方法については一案があった。例えばシクロヘキサンに脱離基が存在する場合、求核置換反応を行うとSN2反応の生成物とともにE2反応でオレフィンが副生物となることが散見される。脱離基はaxialまたはpseudo-axial配座をとってSN2反応が進行するが、隣接する炭素にtrans-Hが存在すればC-H結合とC-脱離基結合のσ*軌道の重なりが大きくなりE2反応が起こりやすくなる。一方、シクロヘキセンオキシド(エポキサイド)のSN2置換反応においては、このような軌道の重なりが小さいためにオレフィンの生成は抑制気味となる(全く生成しないわけではない)。とにかく、この事実は頭の中に入っていたので、シクロプロパン環をCl-で開裂させることを企画した。まず、市販の(+)-carveol (1-1) をマロン酸エステルに変換してジアゾトランスファーにより (1-2) を得た。これを銅触媒 (2-1) 存在下で加熱してシクロプロパン誘導体 (3-2) を得た。(3-2) の環開裂は少し過激な条件を必要としたが、DMF中でLiClとカンファースルホン酸 (CSA) 存在下で140度に加熱することで目的物をまずまずの収率で得ることができた。脱C O2Me基はKrapchoの条件に近いから当然のことである。
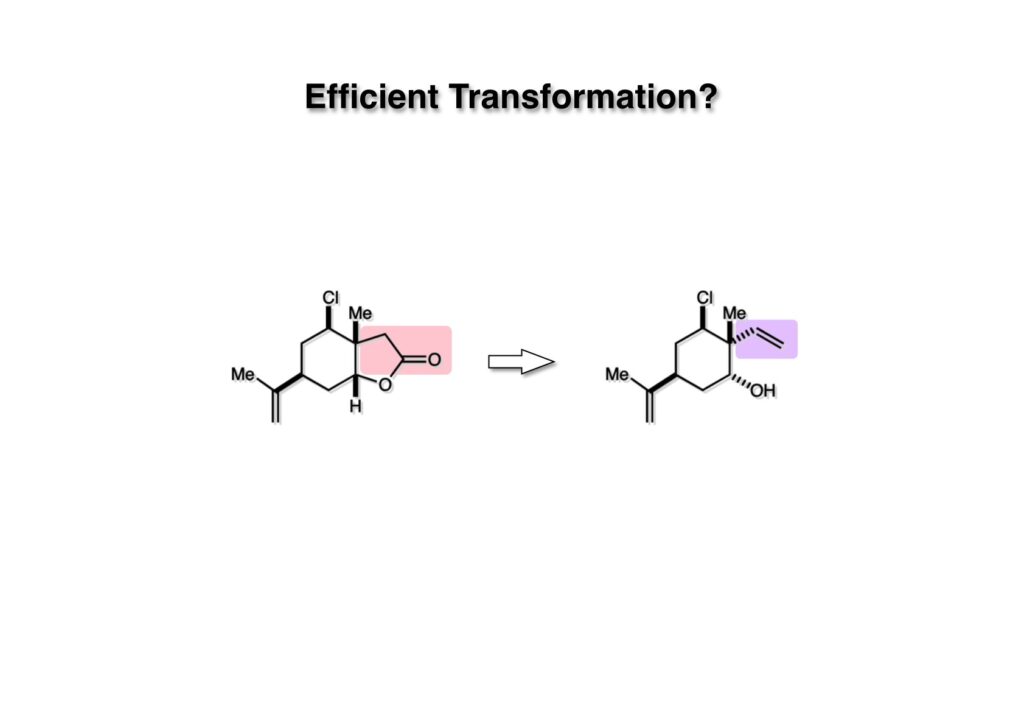 次の課題はラクトン (1-1) の2炭素から (1-2) のビニル基を効率的に構築することだ。
次の課題はラクトン (1-1) の2炭素から (1-2) のビニル基を効率的に構築することだ。
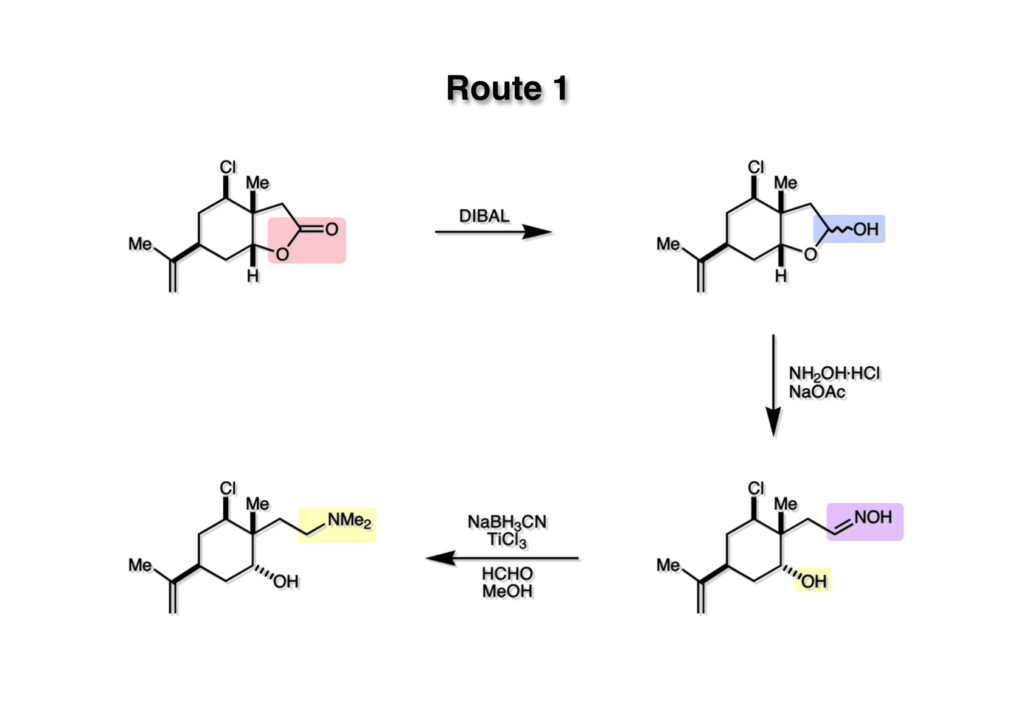 まずラクトン (1-1) をDIBAL還元してラクトール (1-2) に変換した。ラクトールの開環には色々な手段がある。ヒドラジンを使ってヒドラゾンにしたり、NH2OHでオキシムにする方法。EtSHと酸を使ってチオアセタールにするか、安定イリドを使ってWittig反応を行うなどなどである。ここではオキシム (2-2) に変換し、さらにごっちゃ煮みたいにNaBH3CNでオキシムをヒドロキシルアミンにしてからTiCl3でアミンに還元し、ホルムアルデヒドとNaBH3CNで還元的メチル化にするという過程をワンポットで実行してジメチルアミン (2-1) を得た。
まずラクトン (1-1) をDIBAL還元してラクトール (1-2) に変換した。ラクトールの開環には色々な手段がある。ヒドラジンを使ってヒドラゾンにしたり、NH2OHでオキシムにする方法。EtSHと酸を使ってチオアセタールにするか、安定イリドを使ってWittig反応を行うなどなどである。ここではオキシム (2-2) に変換し、さらにごっちゃ煮みたいにNaBH3CNでオキシムをヒドロキシルアミンにしてからTiCl3でアミンに還元し、ホルムアルデヒドとNaBH3CNで還元的メチル化にするという過程をワンポットで実行してジメチルアミン (2-1) を得た。
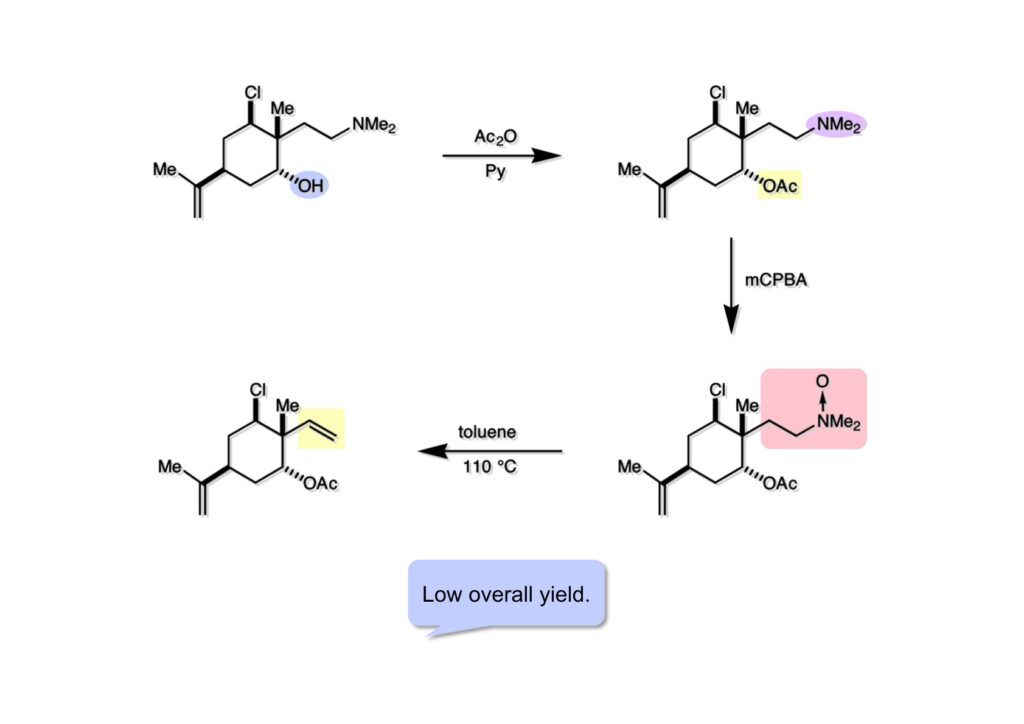 (1-1) の水酸基をアセチル化して保護し、得られた (1-2) をMCPBAで酸化してアミンオキシド (2-2) を得た。これをトルエン中で加熱環流してCope eliminationを実行したが多段階を要することと通算収率が低かったのでこのルートは断念した。また、Cope脱離反応で脱酸素されたアミン (1-2) が少なからず副生したことも却下の原因である。
(1-1) の水酸基をアセチル化して保護し、得られた (1-2) をMCPBAで酸化してアミンオキシド (2-2) を得た。これをトルエン中で加熱環流してCope eliminationを実行したが多段階を要することと通算収率が低かったのでこのルートは断念した。また、Cope脱離反応で脱酸素されたアミン (1-2) が少なからず副生したことも却下の原因である。
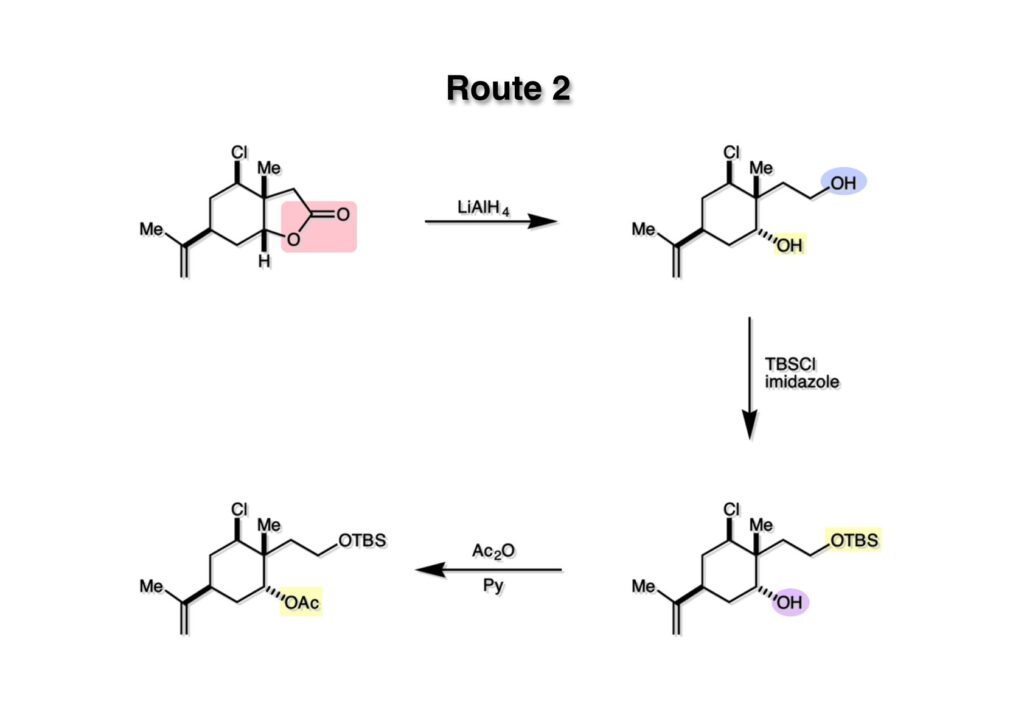 次の試みはselenoxide eliminationを用いたビニル基の構築ルートである。まずラクトン (1-1) をLAH還元してジオール (1-2) に変換した。一級アルコールをTBS基で選択的に保護し、得られた (2-2) の水酸基をアセチル化して (2-1) を得た。
次の試みはselenoxide eliminationを用いたビニル基の構築ルートである。まずラクトン (1-1) をLAH還元してジオール (1-2) に変換した。一級アルコールをTBS基で選択的に保護し、得られた (2-2) の水酸基をアセチル化して (2-1) を得た。
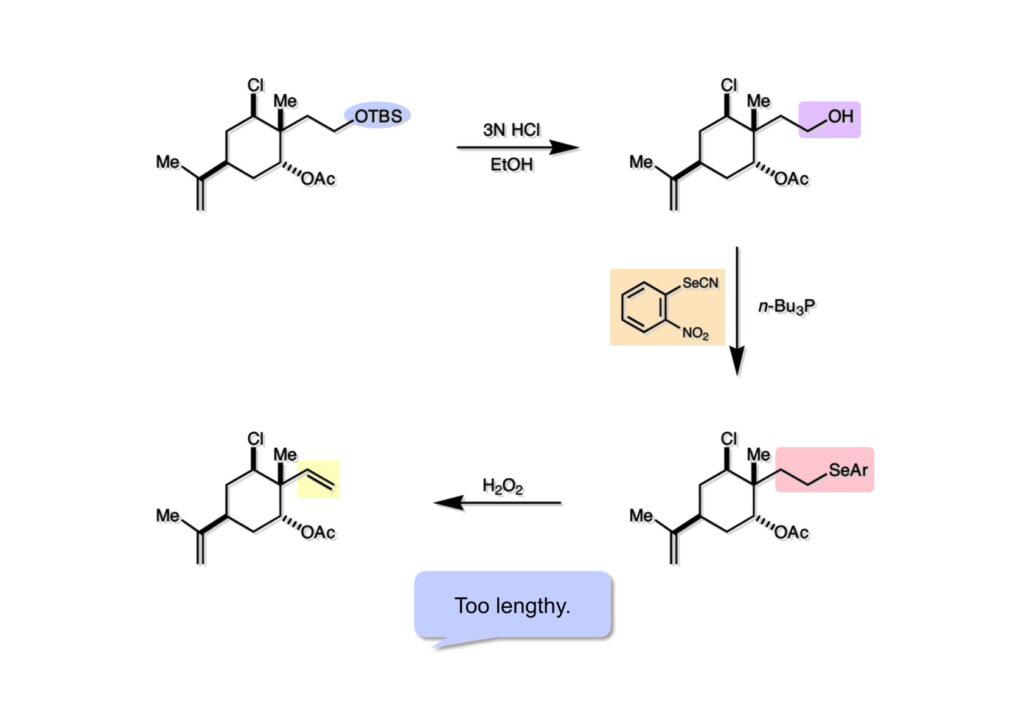 (1-1) のTBS基をTBAFで除去しようとしたら生成したアルコールの方にアセチル基が転位してしまったので希塩酸を使って脱シリル化して (1-2) を得た。アルコール (1-2) からセレナイド (2-2) への変換はGrieco-Nishizawa法を用いた。この反応機構は光延反応と同様である。西沢麦夫さんは若い頃にピッグバーグ大学のPaul Griecoのところでポスドクをし、後に徳島文理大学の教授になられたが日本薬学会賞を受賞されて間もなく若くして亡くなられた。やる気満々の有能で面白い化学者だった。ライス大学で講演された時に拙宅に泊まっていただいたが、西沢さんしか麻雀の点数が数えられず、結局家内や子供達にスカンピンにされた。私は結婚前には麻雀をやったことがなく、家内にやり方を教えてもらったが、上がりが近づくと急に無口になるので皆に丸わかりになった。こういう遊びは苦手である。オルト位にニトロ基を持つセレノキサイドは室温でも脱離が進行する。ここでも (2-2) を過酸加水素で酸化するとすぐに脱離が起きてビニル体 (2-1) が得られた。しかし、このルートの欠点は長すぎることで、こんな手間なんかはかけたくないので別ルートを模索することにした。
(1-1) のTBS基をTBAFで除去しようとしたら生成したアルコールの方にアセチル基が転位してしまったので希塩酸を使って脱シリル化して (1-2) を得た。アルコール (1-2) からセレナイド (2-2) への変換はGrieco-Nishizawa法を用いた。この反応機構は光延反応と同様である。西沢麦夫さんは若い頃にピッグバーグ大学のPaul Griecoのところでポスドクをし、後に徳島文理大学の教授になられたが日本薬学会賞を受賞されて間もなく若くして亡くなられた。やる気満々の有能で面白い化学者だった。ライス大学で講演された時に拙宅に泊まっていただいたが、西沢さんしか麻雀の点数が数えられず、結局家内や子供達にスカンピンにされた。私は結婚前には麻雀をやったことがなく、家内にやり方を教えてもらったが、上がりが近づくと急に無口になるので皆に丸わかりになった。こういう遊びは苦手である。オルト位にニトロ基を持つセレノキサイドは室温でも脱離が進行する。ここでも (2-2) を過酸加水素で酸化するとすぐに脱離が起きてビニル体 (2-1) が得られた。しかし、このルートの欠点は長すぎることで、こんな手間なんかはかけたくないので別ルートを模索することにした。
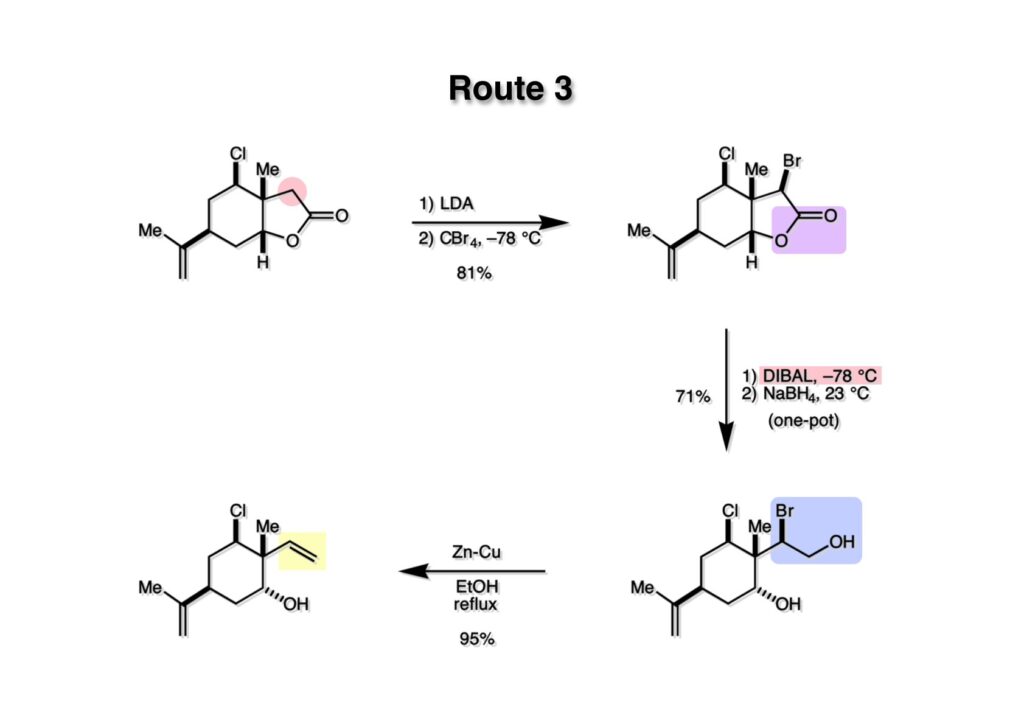 次のアイデアはブロモヒドリンを還元的にオレフィンに変換するルートだ。まずラクトン (1-1) をLDAでカルバニオンにしてCBr4を加えてブルモ化して (1-2) を得た。幸いこの条件下ではシクロプロパンに閉環することなくCl基は無事に残った。ラクトンの還元をLAHやNaBH4で行おうとしたら脱ブロモ化が起きてしまった。これらのate型の還元剤はSET (single electron transfer) が起こりやすいことが知られている。即ち、ラクトンのカルボニル基に1電子が移動すればラジカルアニオンが生成し、そこからBr-が脱離するという反応機構だ。これを避けるためにはLewis酸型の還元剤を使うべきで、この場合DIBALを使うことでBr基を保持したままラクトンがラクトールに還元される。DIBALではそれ以上に還元が進行しないので、ラクトール生成が完了後にメタノールとNaBH4を加えることでジオール (2-2) にまで還元される。ここで水酸基をメシレートにすれば還元が進行しやすいかもしれないが、幸いブロモヒドリンをZn-Cu (zinc copper couple) でエタノール中環流することで高収率で望むビニル体 (2-1) が得られた。
次のアイデアはブロモヒドリンを還元的にオレフィンに変換するルートだ。まずラクトン (1-1) をLDAでカルバニオンにしてCBr4を加えてブルモ化して (1-2) を得た。幸いこの条件下ではシクロプロパンに閉環することなくCl基は無事に残った。ラクトンの還元をLAHやNaBH4で行おうとしたら脱ブロモ化が起きてしまった。これらのate型の還元剤はSET (single electron transfer) が起こりやすいことが知られている。即ち、ラクトンのカルボニル基に1電子が移動すればラジカルアニオンが生成し、そこからBr-が脱離するという反応機構だ。これを避けるためにはLewis酸型の還元剤を使うべきで、この場合DIBALを使うことでBr基を保持したままラクトンがラクトールに還元される。DIBALではそれ以上に還元が進行しないので、ラクトール生成が完了後にメタノールとNaBH4を加えることでジオール (2-2) にまで還元される。ここで水酸基をメシレートにすれば還元が進行しやすいかもしれないが、幸いブロモヒドリンをZn-Cu (zinc copper couple) でエタノール中環流することで高収率で望むビニル体 (2-1) が得られた。
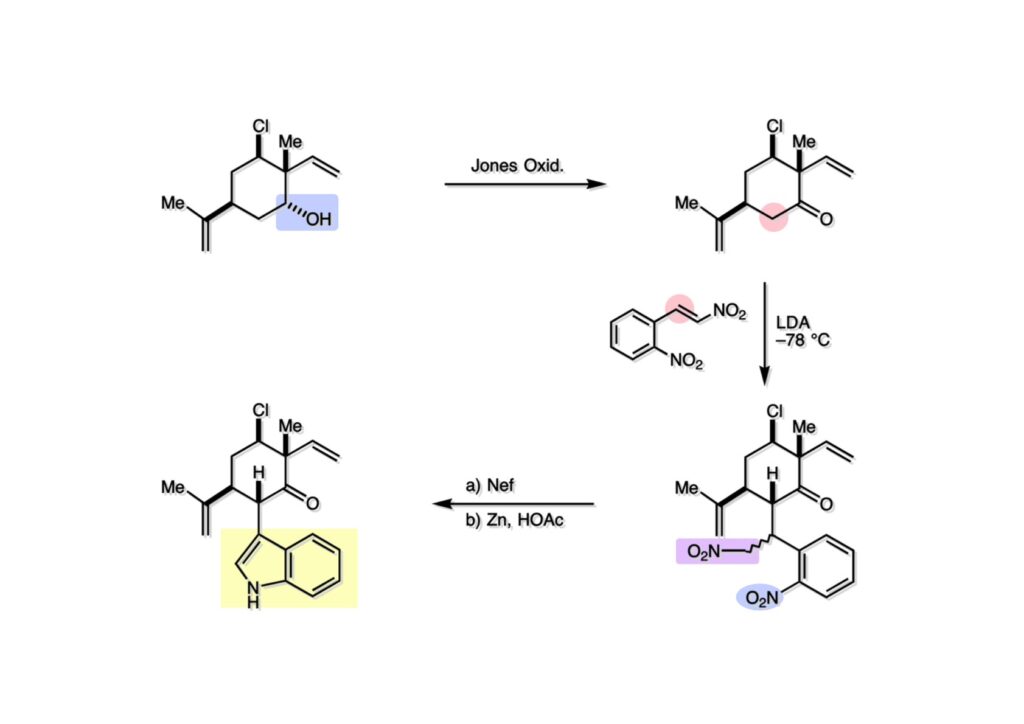 次にインドールの4位にFriedel-Crafts型の置換反応が可能かどうかを確かめるべく駒を進めた。まず、(1-1) の水酸基をJones酸化してケトン (1-2) に変換した。このケトンをLDAで脱プロトン化し、そこにニトロオレフィン (2-1) を加えたところ付加反応が進行してジニトロ体 (3-2) を得た。ニトロアルカンをNef反応でアルデヒドにしてから芳香環上のニトロ基を亜鉛還元すると目的とするインドール (3-1) が得られた。
次にインドールの4位にFriedel-Crafts型の置換反応が可能かどうかを確かめるべく駒を進めた。まず、(1-1) の水酸基をJones酸化してケトン (1-2) に変換した。このケトンをLDAで脱プロトン化し、そこにニトロオレフィン (2-1) を加えたところ付加反応が進行してジニトロ体 (3-2) を得た。ニトロアルカンをNef反応でアルデヒドにしてから芳香環上のニトロ基を亜鉛還元すると目的とするインドール (3-1) が得られた。
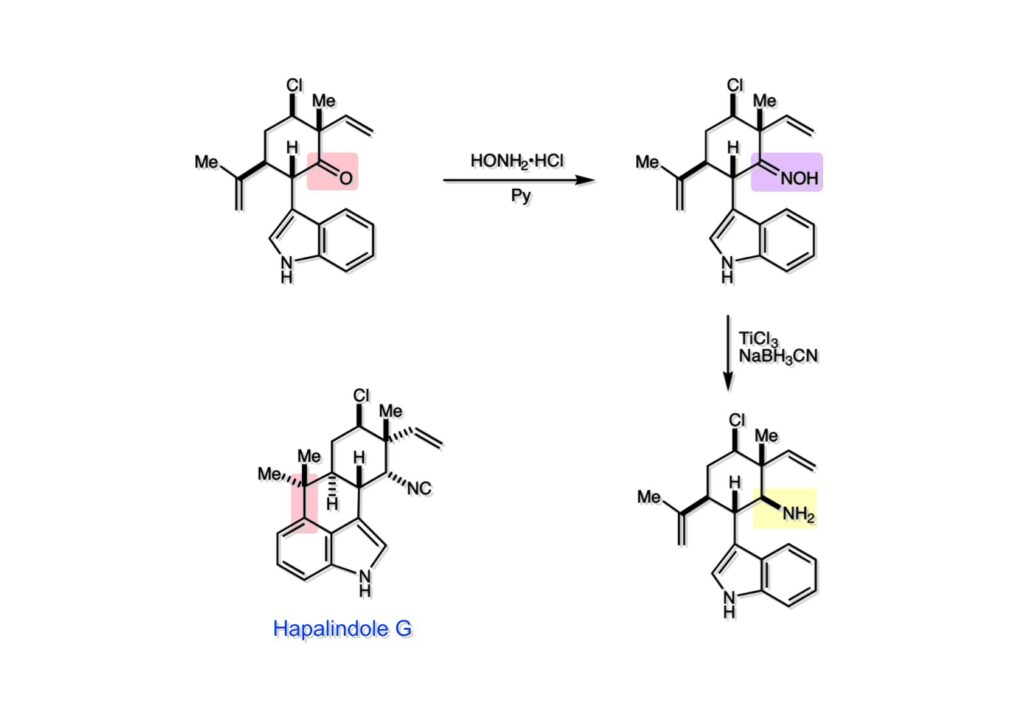 ケトン (1-1) は還元的アミノ化が進行しなかったので、まずオキシム (1-2) に変換してからTiCl3-NaBH3CNの条件でアミン (2-2) が得られたが、NMR解析によってアミノ基はβ配置であると判明した。
ケトン (1-1) は還元的アミノ化が進行しなかったので、まずオキシム (1-2) に変換してからTiCl3-NaBH3CNの条件でアミン (2-2) が得られたが、NMR解析によってアミノ基はβ配置であると判明した。
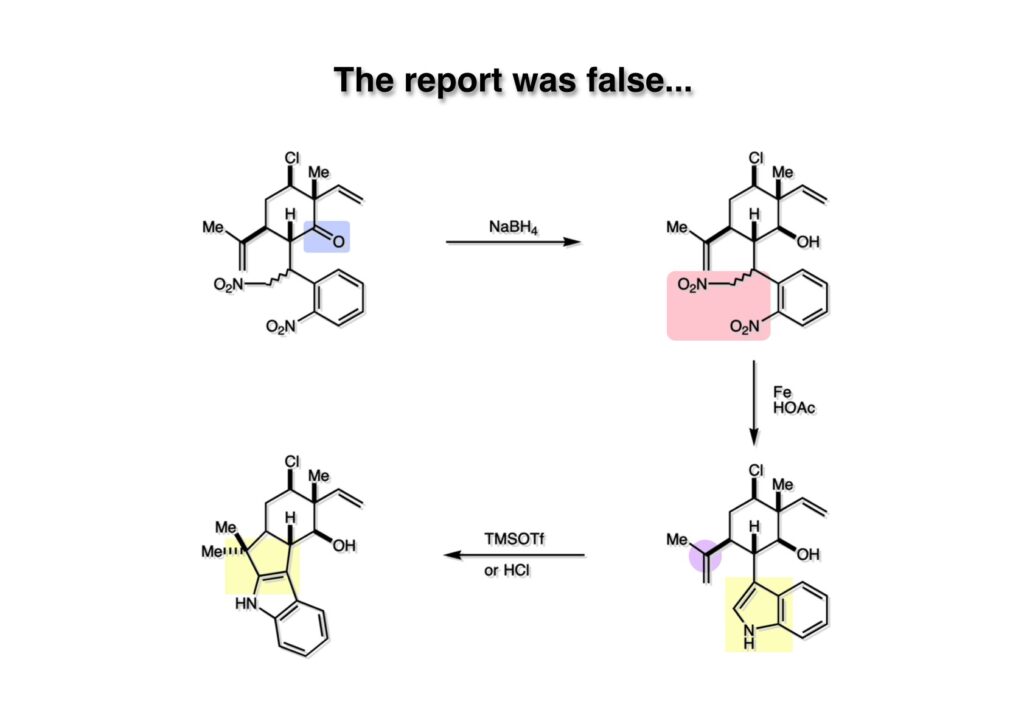 Dick Mooreと元ポスドクが (2-2) に類似の天然物 (hapalindole E formamide) をEtOH中希塩酸で処理するとhapalindole G amineが得られるとJOCに報告した。エッ!そんな反応が本当に起きるのか?と信じられなかったが、取り敢えずOH体 (2-2) を合成してTMSOTfや希塩酸と加熱してみたが得られるのはインドールの2位に置換した化合物 (2-1) のみであった。Moore研での化合物は我々が行ったβ配置のアルコールでなくα配置のフォルムアミドだったが、そんなに劇的な違いが生ずるのかは極めて疑わしい。まあ、同一化合物での実験でないので100%間違っているとは言えないけど。
Dick Mooreと元ポスドクが (2-2) に類似の天然物 (hapalindole E formamide) をEtOH中希塩酸で処理するとhapalindole G amineが得られるとJOCに報告した。エッ!そんな反応が本当に起きるのか?と信じられなかったが、取り敢えずOH体 (2-2) を合成してTMSOTfや希塩酸と加熱してみたが得られるのはインドールの2位に置換した化合物 (2-1) のみであった。Moore研での化合物は我々が行ったβ配置のアルコールでなくα配置のフォルムアミドだったが、そんなに劇的な違いが生ずるのかは極めて疑わしい。まあ、同一化合物での実験でないので100%間違っているとは言えないけど。
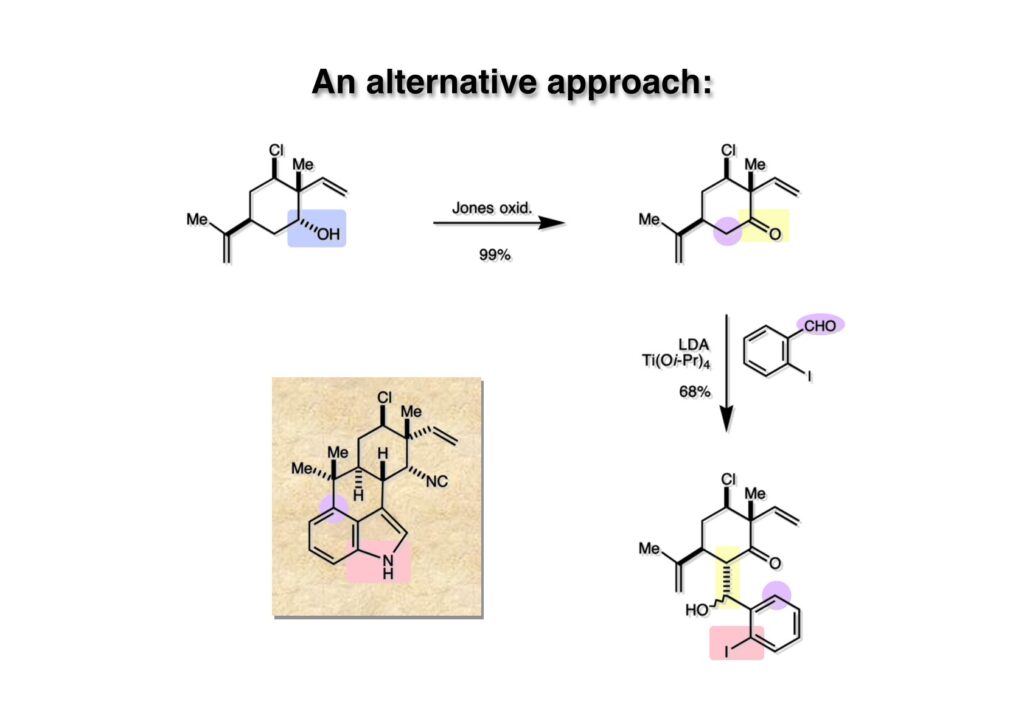 インドール環を構築してからFriedel-Crafts反応を行うことは勝算が無さそうだったので、後でインドールに変換可能な中間体で実行することにした。キトン (1-2) をLDAで脱プロトン化してからo-iodobenzaldehyde (2-1) を加えたところ、予期に反して付加体は全く得られなかった。アルデヒドが還元されてo-iodobenzyl alcoholが得られて、ケトン (1-2) は回収された。こんな経験は初めてで、何らかの電子移動が関与していると思われる。そこでエノレートの反応性を抑えて、Lewis酸性のより高いチタン (Ti(Oi-Pr)4))を使うアルドール反応がManfred Reetzによって報告されていたので本反応に適用したところ目的とする付加体 (3-1) がジアステレオマー混合物として得られた。
インドール環を構築してからFriedel-Crafts反応を行うことは勝算が無さそうだったので、後でインドールに変換可能な中間体で実行することにした。キトン (1-2) をLDAで脱プロトン化してからo-iodobenzaldehyde (2-1) を加えたところ、予期に反して付加体は全く得られなかった。アルデヒドが還元されてo-iodobenzyl alcoholが得られて、ケトン (1-2) は回収された。こんな経験は初めてで、何らかの電子移動が関与していると思われる。そこでエノレートの反応性を抑えて、Lewis酸性のより高いチタン (Ti(Oi-Pr)4))を使うアルドール反応がManfred Reetzによって報告されていたので本反応に適用したところ目的とする付加体 (3-1) がジアステレオマー混合物として得られた。
 Friedel-Crafts反応を実行するために (1-1) の水酸基をアセチル化してDBUで酢酸を脱離させるとα,β-不飽和ケトン (1-2) を得た。単一物が生成したが二重結合の配置は決定していない。オルト位にヨウ素が入れてあるのには勿論理由がある。この位置にN3やNH2、それにNO2基を入れておくと環化が起きないことが分かっていた。一方、Hのままで酸性条件に付すと環化は進行した。そこで、ベンゼン環の電子密度をなるべくフェニル基に近いもので、尚且つ窒素に変換可能なものは何かということを考えるために電気陰性度の値に注目した。H (2.20), C (2.55), N (3.04), Cl (3.16), Br (2.96), I (2.66) というようにヨウ素の電気陰性度はかなり炭素に近いことが分かる。確かに (1-2) をメタンスルホン酸-TFA (1:10) の混合溶液中で室温放置すると高収率で目的とする環化化合物 (2-2) が得られた。次にパラジウム触媒を用いてCOと反応させるとカルボニル化が進行してカルボン酸 (2-1) が得られた。
Friedel-Crafts反応を実行するために (1-1) の水酸基をアセチル化してDBUで酢酸を脱離させるとα,β-不飽和ケトン (1-2) を得た。単一物が生成したが二重結合の配置は決定していない。オルト位にヨウ素が入れてあるのには勿論理由がある。この位置にN3やNH2、それにNO2基を入れておくと環化が起きないことが分かっていた。一方、Hのままで酸性条件に付すと環化は進行した。そこで、ベンゼン環の電子密度をなるべくフェニル基に近いもので、尚且つ窒素に変換可能なものは何かということを考えるために電気陰性度の値に注目した。H (2.20), C (2.55), N (3.04), Cl (3.16), Br (2.96), I (2.66) というようにヨウ素の電気陰性度はかなり炭素に近いことが分かる。確かに (1-2) をメタンスルホン酸-TFA (1:10) の混合溶液中で室温放置すると高収率で目的とする環化化合物 (2-2) が得られた。次にパラジウム触媒を用いてCOと反応させるとカルボニル化が進行してカルボン酸 (2-1) が得られた。
 カルボン酸 (1-1) のCurtius転位は塩入先生の開発されたDPPAとアリルアルコール存在下で加熱してAlloc基で保護されたアミン (1-2) に変換した。(1-2) からインドールを構築するにはアルデヒドを共役付加する必要がある。幸いこのケトンにはαプロトンが無いのでMMTMS (FAMSO) のリチウム塩 (2-1) を直接付加させることができる。得られた付加体 (3-2) を精製することなくHClO4と加熱することによりN-Allocインドール (3-1) が得られた。ここでHgCl2を加えてあるのは万が一MeSHが遊離した場合にトラップするためである。
カルボン酸 (1-1) のCurtius転位は塩入先生の開発されたDPPAとアリルアルコール存在下で加熱してAlloc基で保護されたアミン (1-2) に変換した。(1-2) からインドールを構築するにはアルデヒドを共役付加する必要がある。幸いこのケトンにはαプロトンが無いのでMMTMS (FAMSO) のリチウム塩 (2-1) を直接付加させることができる。得られた付加体 (3-2) を精製することなくHClO4と加熱することによりN-Allocインドール (3-1) が得られた。ここでHgCl2を加えてあるのは万が一MeSHが遊離した場合にトラップするためである。
 ケトン (1-1) の還元はNaBH4のaxial攻撃によってβ配置のアルコール (1-2) が生成する。この混み合った水酸基のメシル化はメシルクロライドでは役不足で、メタンスルホン酸無水物を使わなければならない。これは岸研でマイトマイシンCの合成をやっていた時にMsClでメシル化が出来なかった時に解決したのがMs2Oだったという経験からである。さて、この立体障害の大きなメシレート (2-2) のLiN3によるSN2反応は予想以上に困難だったが、隣接する炭素に結合した水素がメシレートに対してシス配置だったのが幸いした。トランスだったら置換でなく脱離が起きていただろう。100度36時間という条件ながらも高収率でアジド体 (2-1) が得られた。
ケトン (1-1) の還元はNaBH4のaxial攻撃によってβ配置のアルコール (1-2) が生成する。この混み合った水酸基のメシル化はメシルクロライドでは役不足で、メタンスルホン酸無水物を使わなければならない。これは岸研でマイトマイシンCの合成をやっていた時にMsClでメシル化が出来なかった時に解決したのがMs2Oだったという経験からである。さて、この立体障害の大きなメシレート (2-2) のLiN3によるSN2反応は予想以上に困難だったが、隣接する炭素に結合した水素がメシレートに対してシス配置だったのが幸いした。トランスだったら置換でなく脱離が起きていただろう。100度36時間という条件ながらも高収率でアジド体 (2-1) が得られた。
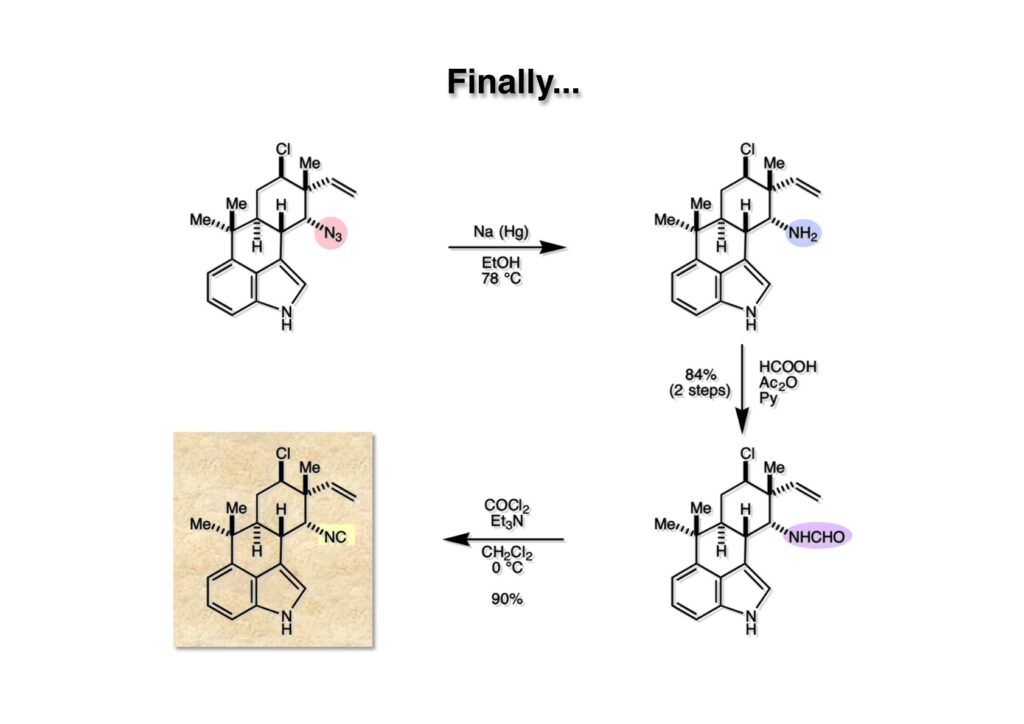 このアジド (1-1) が亜鉛-酢酸でアミンに還元できなかったのには驚いた。本当にアジドなんだろうか?と疑問に思ったくらいだ。そこでより強い還元力のあるナトリウムアマルガム (Na-Hg) を用いてエタノール中で加熱したところ幸いにもアミン (1-2) が得られた。Na-Hgはナトリウムと水銀を混ぜることで簡単に出来上がるが、石ころみたいに硬くてXiaoqiが金槌でガンガン叩いて小さくしたのを覚えている。アミン、ギ酸、ピリジンの溶液に少しずつ無水酢酸を加えていくとホルムアミド体 (2-2) が得られる。それをホスゲンで脱水するとhapalindole Gが得られた。スペクトルは報告されたものと一致したが、Dick Mooreのところにサンプルを送って天然物に一致することを確かめてもらった。もっと短工程の合成にしたかったが、なかなか難しい化合物だったと言える。
このアジド (1-1) が亜鉛-酢酸でアミンに還元できなかったのには驚いた。本当にアジドなんだろうか?と疑問に思ったくらいだ。そこでより強い還元力のあるナトリウムアマルガム (Na-Hg) を用いてエタノール中で加熱したところ幸いにもアミン (1-2) が得られた。Na-Hgはナトリウムと水銀を混ぜることで簡単に出来上がるが、石ころみたいに硬くてXiaoqiが金槌でガンガン叩いて小さくしたのを覚えている。アミン、ギ酸、ピリジンの溶液に少しずつ無水酢酸を加えていくとホルムアミド体 (2-2) が得られる。それをホスゲンで脱水するとhapalindole Gが得られた。スペクトルは報告されたものと一致したが、Dick Mooreのところにサンプルを送って天然物に一致することを確かめてもらった。もっと短工程の合成にしたかったが、なかなか難しい化合物だったと言える。