協和発酵の平田正さん(後に社長)から夜遅く自宅に電話があり、面白い化合物があるから全合成をやってみないかという事だった。平田さんが東京研究所の研究管理室長だった時に「マイトマイシンCの全合成」の講演をして以来のお付き合いで、若い優秀な研究員を送るからということと、硫黄が3つ入っていて非常にユニークな化合物と説明された。ただ、構造式のイメージが湧いてこなかったのでオフィスにファクスで構造式を送って欲しいと依頼した。翌日オフィスに行き構造式を見た瞬間に全合成をやろうと即決した。送られた北神田裕さんは東工大の辻研修士出身で高橋孝志さんの弟子だった。非常に優秀で、実験が上手なことは言うまでもないが、必要な情報などを図書館から探すのに彼ほど速くて正確な学生・研究者は見たことが無かった。ライス大学で2年ちょっと、帰国して1年ほど、計3年余りの期間に単独で全合成を完成させた。日本に帰国してからはファクスで頻繁に連絡を取り合って研究を進めた。何しろ手探り状態で色々なアプローチを行ったが、難易度に関しては当研究室で合成した化合物の中でトップ3には確実に入るだろう。
“Total Synthesis of (+)-Leinamycin,” Y. Kanda and T. Fukuyama, J. Am. Chem. Soc., 115, 8451 (1993).
“Total Synthesis of (+)-Leinamycin,” T. Fukuyama and Y. Kanda, J. Synth. Org. Chem., Jpn., 52, 888 (1994).
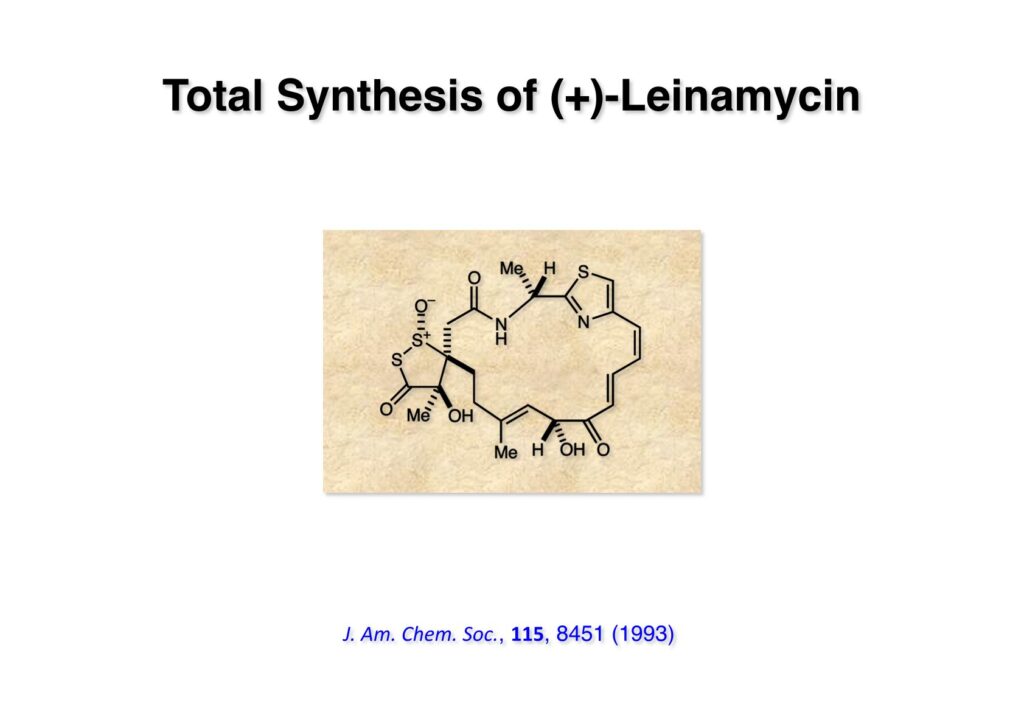 Leinamycinは抗腫瘍活性があり、上市が期待されてプロドラッグ化など協和発酵で精力的に開発研究が為されたが、結局薬にはならなかった。スピロ環を持つ大環状ラクタムで特に左の硫黄原子2個を含む5員環は天然物としては類を見ない構造で「One of a Kind」と言う表現がピッタリである。私たちの他に1グループが予備実験的なアプローチを報告しているが、おそらく本気で全合成しようとは思っていなかったのだろう。現在までに全合成に成功したのは当研究室だけである。
Leinamycinは抗腫瘍活性があり、上市が期待されてプロドラッグ化など協和発酵で精力的に開発研究が為されたが、結局薬にはならなかった。スピロ環を持つ大環状ラクタムで特に左の硫黄原子2個を含む5員環は天然物としては類を見ない構造で「One of a Kind」と言う表現がピッタリである。私たちの他に1グループが予備実験的なアプローチを報告しているが、おそらく本気で全合成しようとは思っていなかったのだろう。現在までに全合成に成功したのは当研究室だけである。
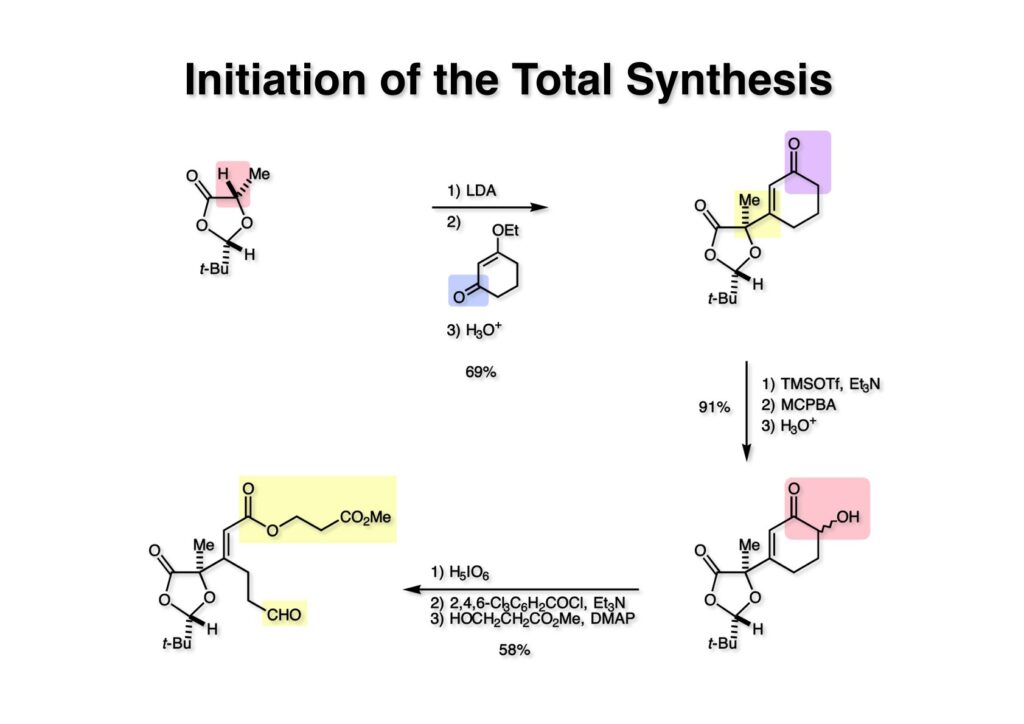
 Leinamycin (1-1) はピリジンと水存在下で室温で徐々に分解するそうで、一番不安定そうな5員環の構築は合成の終盤に行うことにした。α,β-不飽和ラクタム (1-2) に何とか分子内反応で硫黄をMichael付加すれば良いのではないか、と合成開始当初は問題を先送りしておいた。(1-2) のラクタムは当然開裂させておき、次に注目したのはcis–transのジエン構造だった。このcis-オレフィンは三重結合の部分還元を行えば良いので、薗頭カップリングを念頭に置いてチアゾール体 (2-3) とtransのハロオレフィン (2-2) の2分子に簡略化した。α-Hydroxyketone (2-2) の二級水酸基の立体化学はα-hydroxycarboxylic acidの立体化学を利用するには離れすぎているので、何らかの不斉アルドール反応を利用すべくアルデヒド (2-1) に導いた。
Leinamycin (1-1) はピリジンと水存在下で室温で徐々に分解するそうで、一番不安定そうな5員環の構築は合成の終盤に行うことにした。α,β-不飽和ラクタム (1-2) に何とか分子内反応で硫黄をMichael付加すれば良いのではないか、と合成開始当初は問題を先送りしておいた。(1-2) のラクタムは当然開裂させておき、次に注目したのはcis–transのジエン構造だった。このcis-オレフィンは三重結合の部分還元を行えば良いので、薗頭カップリングを念頭に置いてチアゾール体 (2-3) とtransのハロオレフィン (2-2) の2分子に簡略化した。α-Hydroxyketone (2-2) の二級水酸基の立体化学はα-hydroxycarboxylic acidの立体化学を利用するには離れすぎているので、何らかの不斉アルドール反応を利用すべくアルデヒド (2-1) に導いた。
 (1-1) の不飽和アルデヒドの構築はメチルケトンからHorner-Emmons反応などで2炭素増炭するつもりであったが、出発物をより簡素化するためには (1-2) のようなカルボン酸から構築することも可能だと考えた。ここをカルボン酸にすることで、不飽和カルボン酸とのC-C結合形成により3置換オレフィンの立体化学をシクロへキセノン (2-2) のように規定することができるからである。さて、最後の不斉炭素の構築であるが、Dieter Seebach教授の光学活性ジオキソラノン (1-1) に頼らざるを得なかった。
(1-1) の不飽和アルデヒドの構築はメチルケトンからHorner-Emmons反応などで2炭素増炭するつもりであったが、出発物をより簡素化するためには (1-2) のようなカルボン酸から構築することも可能だと考えた。ここをカルボン酸にすることで、不飽和カルボン酸とのC-C結合形成により3置換オレフィンの立体化学をシクロへキセノン (2-2) のように規定することができるからである。さて、最後の不斉炭素の構築であるが、Dieter Seebach教授の光学活性ジオキソラノン (1-1) に頼らざるを得なかった。
 (2S,5S)-2-tert-Butyl-5-methyl-1,3-dioxolan-4-one (1-1) はL-lactic acidとpivalaldehydeを酸触媒存在下で脱水縮合してcis体とtrans体の混合物として得られる。これをn-pentaneに溶かしてDry Ice-アセトン浴で冷却するとcis体が結晶となって析出してくる。Seebach研はさすがにスイスの名門大学ETH、Dry Iceで冷却したアセトンがグラスフィルターの外套部分を流れて、濾取した結晶が融けないように単離していた。そんな高級なグラスフィルターなど見たこともなく、貧乏研究室でも高純度の (1-1) が得られるように工夫してみた。アメリカではgas dispersion tube(ガス分散管;ガラス管の先っぽがグラスフィルターみたいなフリットになっている)と言うのが簡単に手に入るので、低温で結晶化した母液を吸引瓶と水流ポンプで吸い出した。この操作を2回繰り返すと純度の高い cis体 (1-1) が得られた。(1-1) をLDAで脱プロトン化し3-ethoxy-2-hexen-1-one (1-2) を加えるとエノンへの 1,2-付加が進行し、これを希塩酸で加水分解するとシクロへキセノン (1-3) が得られた。この方法は3位が置換されたシクロへキセノンを大量に合成するのに便利な反応である。シクロへキセノン (1-3) の6位に水酸基を導入する方法は、まず速度論的に有利な視リルエノールエーテルを作りRubottom酸化を行うと生じたエポキサイドは直ちに開裂してα-hydroxyketoneのシリルエーテルが生成する。これを希塩酸で加水分解して (2-2) を得た。このヒドロキシケトンは過ヨウ素酸で容易に開裂してカルボン酸-アルデヒドが生成する。モデル実験ではカルボン酸をメチルエステルに変換してマクロラクタム形成直前まで進んだが、ジオキソラノン環を保持したままメチルエステルの加水分解が出来なかったので、本番ではこの時点で山口法を使ってカルボン酸とmethyl β-hydroxypropionateとを縮合して (2-1) に変換した。メチルエステルがモデル実験で他段階を生き残ったので、同様のメチルエステルでretro-Michael反応でカルボン酸に変換可能な保護基を使ったわけだ。
(2S,5S)-2-tert-Butyl-5-methyl-1,3-dioxolan-4-one (1-1) はL-lactic acidとpivalaldehydeを酸触媒存在下で脱水縮合してcis体とtrans体の混合物として得られる。これをn-pentaneに溶かしてDry Ice-アセトン浴で冷却するとcis体が結晶となって析出してくる。Seebach研はさすがにスイスの名門大学ETH、Dry Iceで冷却したアセトンがグラスフィルターの外套部分を流れて、濾取した結晶が融けないように単離していた。そんな高級なグラスフィルターなど見たこともなく、貧乏研究室でも高純度の (1-1) が得られるように工夫してみた。アメリカではgas dispersion tube(ガス分散管;ガラス管の先っぽがグラスフィルターみたいなフリットになっている)と言うのが簡単に手に入るので、低温で結晶化した母液を吸引瓶と水流ポンプで吸い出した。この操作を2回繰り返すと純度の高い cis体 (1-1) が得られた。(1-1) をLDAで脱プロトン化し3-ethoxy-2-hexen-1-one (1-2) を加えるとエノンへの 1,2-付加が進行し、これを希塩酸で加水分解するとシクロへキセノン (1-3) が得られた。この方法は3位が置換されたシクロへキセノンを大量に合成するのに便利な反応である。シクロへキセノン (1-3) の6位に水酸基を導入する方法は、まず速度論的に有利な視リルエノールエーテルを作りRubottom酸化を行うと生じたエポキサイドは直ちに開裂してα-hydroxyketoneのシリルエーテルが生成する。これを希塩酸で加水分解して (2-2) を得た。このヒドロキシケトンは過ヨウ素酸で容易に開裂してカルボン酸-アルデヒドが生成する。モデル実験ではカルボン酸をメチルエステルに変換してマクロラクタム形成直前まで進んだが、ジオキソラノン環を保持したままメチルエステルの加水分解が出来なかったので、本番ではこの時点で山口法を使ってカルボン酸とmethyl β-hydroxypropionateとを縮合して (2-1) に変換した。メチルエステルがモデル実験で他段階を生き残ったので、同様のメチルエステルでretro-Michael反応でカルボン酸に変換可能な保護基を使ったわけだ。
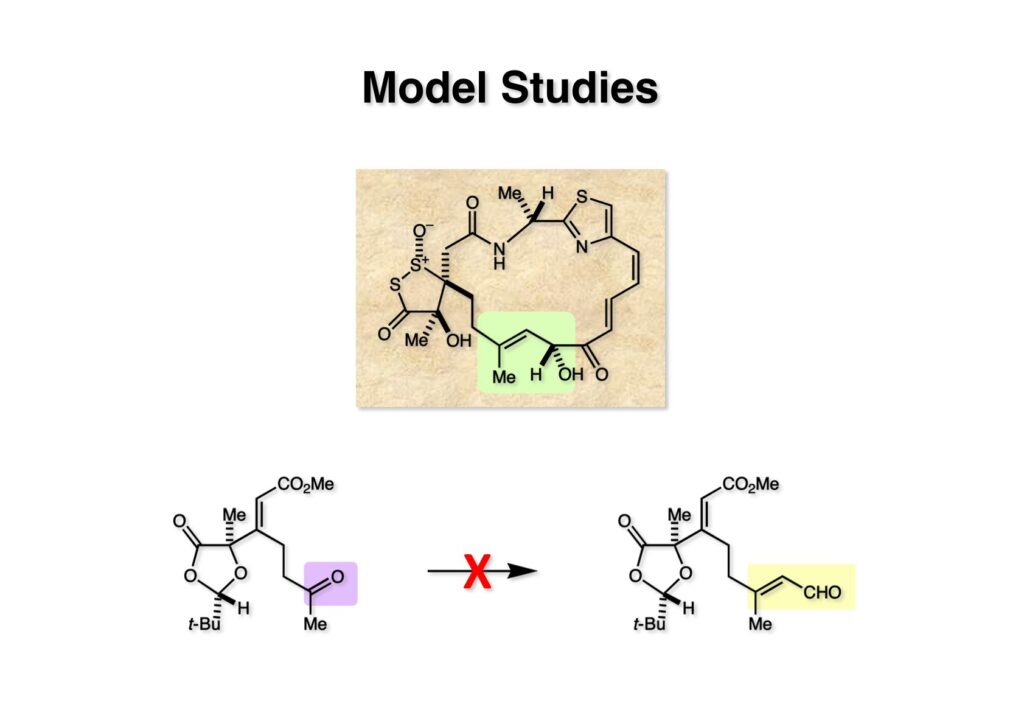 モデル実験で初めに試みたのはメチルケトン (2-1) からの2炭素増炭だった。Wittig反応やHorner-Emmons反応では目的物(エステル)が得られず、アセチレンの付加もジオキソラノンのカルボニル基の反応性がケトンと同じくらいで失敗に終わった。
モデル実験で初めに試みたのはメチルケトン (2-1) からの2炭素増炭だった。Wittig反応やHorner-Emmons反応では目的物(エステル)が得られず、アセチレンの付加もジオキソラノンのカルボニル基の反応性がケトンと同じくらいで失敗に終わった。
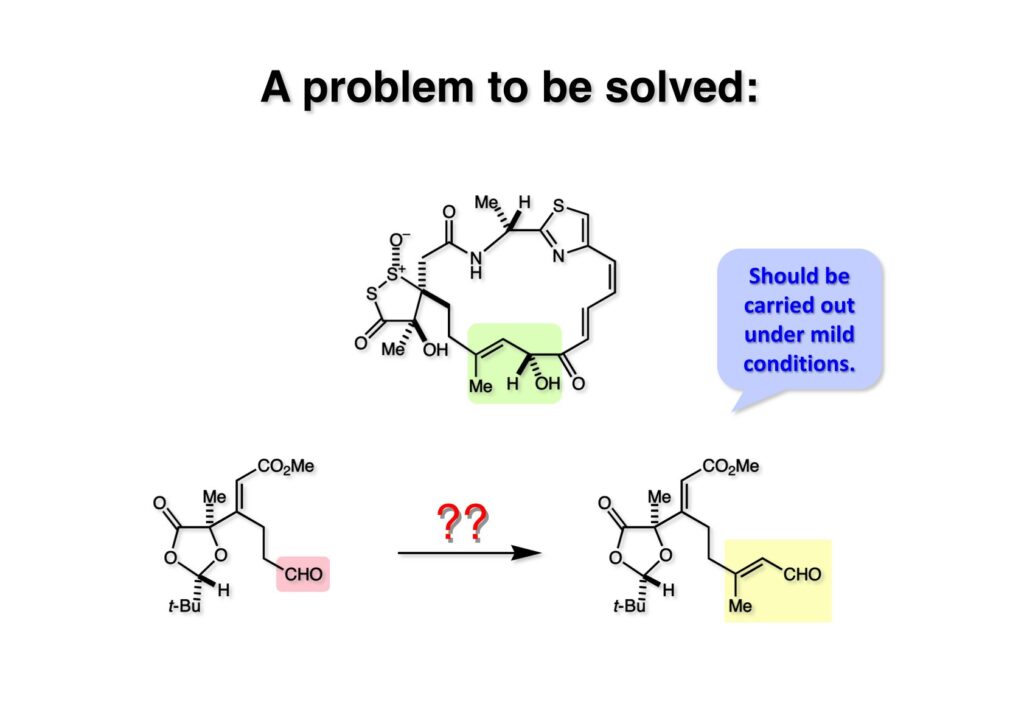 さて、次に可能性を検討したのは (2-1) のようなアルデヒドか、それを酸化して得られるカルボン酸から (2-2) へ変換する方法だ。
さて、次に可能性を検討したのは (2-1) のようなアルデヒドか、それを酸化して得られるカルボン酸から (2-2) へ変換する方法だ。
 ライス大学化学科の院生にcumulative examinationの反応機構問題を出そうとした時に、Organic Synthesesをペラペラと目を通していた。そこで面白いなと思ったのが酸クロライド (1-1) と安定イリド (1-2) の混合物にEt3Nを加えると生成するケテンにWittig反応が進行して (1-3) のようなアレンエステルが生成する反応だった。この反応は記憶に残っていて、アレンエステルのC-3位は非常に反応性が高く共役不可反応が起こりやすいことも常識として知っていた。下のMe2CuLiが (2-1) に付加して (2-2) が得られるという反応は全合成の論文を書くために参考文献として調べただけで、この反応が起こるのは当然だと思っていた。とにかく、(2-2) のような化合物のエステルをアルデヒドにして二重結合を共役させれば目的に叶うのである。
ライス大学化学科の院生にcumulative examinationの反応機構問題を出そうとした時に、Organic Synthesesをペラペラと目を通していた。そこで面白いなと思ったのが酸クロライド (1-1) と安定イリド (1-2) の混合物にEt3Nを加えると生成するケテンにWittig反応が進行して (1-3) のようなアレンエステルが生成する反応だった。この反応は記憶に残っていて、アレンエステルのC-3位は非常に反応性が高く共役不可反応が起こりやすいことも常識として知っていた。下のMe2CuLiが (2-1) に付加して (2-2) が得られるという反応は全合成の論文を書くために参考文献として調べただけで、この反応が起こるのは当然だと思っていた。とにかく、(2-2) のような化合物のエステルをアルデヒドにして二重結合を共役させれば目的に叶うのである。
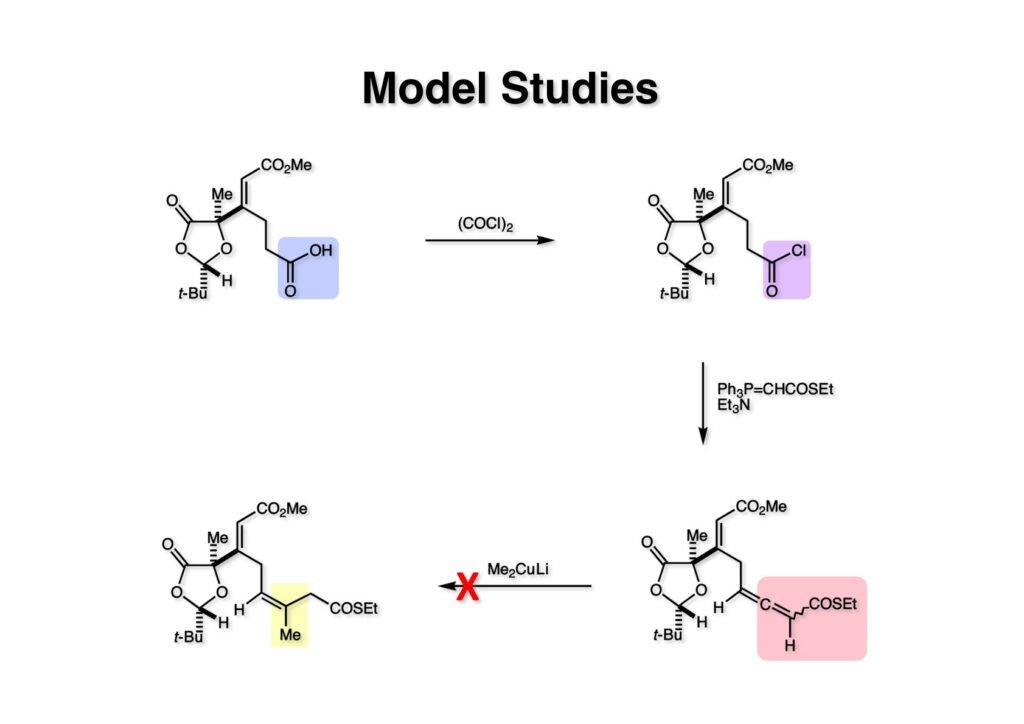 アルデヒドをJones酸化して得たカルボン酸 (1-1) をoxalyl chlorideで酸クロライド (1-2) に変換し、さらにチオエステルを持つ安定イリド (2-1) との混合物にEt3Nを加えると目的とするアレンチオエステル (3-2) がジアステレオマー混合物として得られた。学生諸君はお分かりのようにアレンにはキラル中心が存在するのでジアステレオマーの混合物になるのである。既により簡単な構造を持つアレンチオエステルにMe2CuLiを加えると共役付加が起きることは確認済みで、(3-2) にMe2CuLiを作用させれば当然付加体 (3-1) が生成すると思っていた。ところが、反応を行ってみると (3-1) は全く得られなかった!
アルデヒドをJones酸化して得たカルボン酸 (1-1) をoxalyl chlorideで酸クロライド (1-2) に変換し、さらにチオエステルを持つ安定イリド (2-1) との混合物にEt3Nを加えると目的とするアレンチオエステル (3-2) がジアステレオマー混合物として得られた。学生諸君はお分かりのようにアレンにはキラル中心が存在するのでジアステレオマーの混合物になるのである。既により簡単な構造を持つアレンチオエステルにMe2CuLiを加えると共役付加が起きることは確認済みで、(3-2) にMe2CuLiを作用させれば当然付加体 (3-1) が生成すると思っていた。ところが、反応を行ってみると (3-1) は全く得られなかった!
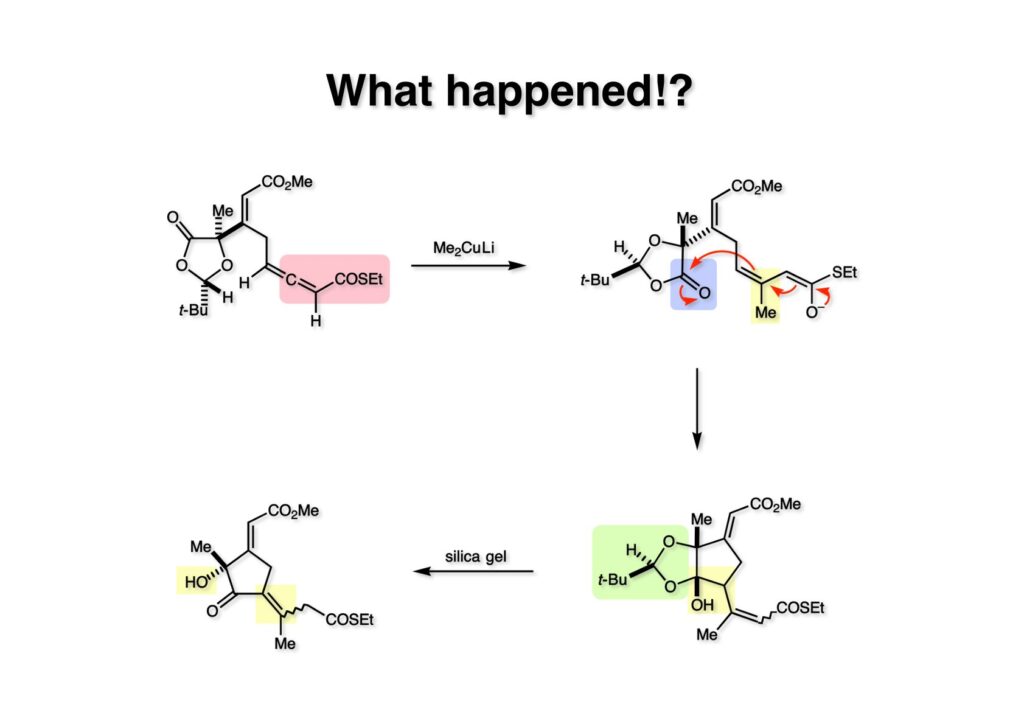 一体何が起こったのか? (1-1) へのMe2CuLiの付加反応は進行したものの、生じたジエノレート (1-2) は低温下直ちにジオキソラノンのカルボニル基を攻撃して2環性の化合物 (2-2) を生じた。反応は非常に綺麗だったので精製せずにNMRを測定したところ如何にも不安定そうなアセタールが残ったままであると判明した。勿論、この化合物は全合成には何の役にも立たない。NMR測定後にシリカゲルで分離したところ (2-1) の混合物が得られた。ここで解決すべき問題は分子内アルドール型付加反応 (1-2) を阻止することだ。
一体何が起こったのか? (1-1) へのMe2CuLiの付加反応は進行したものの、生じたジエノレート (1-2) は低温下直ちにジオキソラノンのカルボニル基を攻撃して2環性の化合物 (2-2) を生じた。反応は非常に綺麗だったので精製せずにNMRを測定したところ如何にも不安定そうなアセタールが残ったままであると判明した。勿論、この化合物は全合成には何の役にも立たない。NMR測定後にシリカゲルで分離したところ (2-1) の混合物が得られた。ここで解決すべき問題は分子内アルドール型付加反応 (1-2) を阻止することだ。
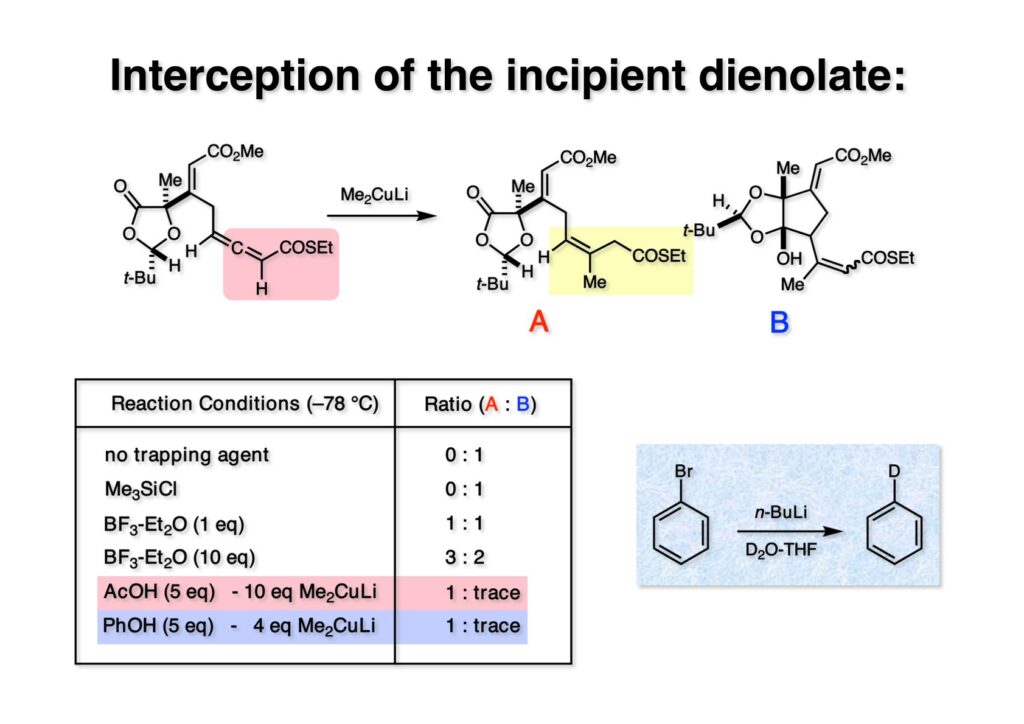 例えばシクロヘキサノンのしリルエノールエーテルを合成するにはケトンとMe3SiClを共存させておいてLDAを加えると目的物が得られるが、(1-1) と過剰のMe3SiClの共存下にMe2CuLiを加えても目的物A (1-2) は全く得られず、不要なB (1-3) のみが得られた。そこで1当量のBF3•Et2O存在下に反応を行ったところ初めて目的物Aと不要物Bが1:1の比で得られてきた。これはいいぞと思い10当量のBF3•Et2Oを用いたところ、わずかに目的物が増えたのみだった。ここで色々考えたのだが、「窮鼠猫を噛む」の例え通り、酢酸存在下で反応を行うことにした。これはただの勘なのだが、エノレートをquenchする最速の種はプロトンであると思ったことと、院生の頃に化学科の図書館でChemistry & Industryという英国で隔週出版されたHighlightsの中にブロモベンゼンとD2O共存下にBuLiを加えるとD化されたベンゼンが生成するという驚き記事を見た記憶があったからだ。おそらくBuLiからPhBrへの電子移動の方がD2Oへのそれより速いということだろうが、Me2CuLiの反応機構も電子移動が先行するという報告を見たことがあった。神田さんは私の無茶なリクエストに驚いただろうが、一応理屈だけはうまくいくかも知れないという期待はあった。果たして、5当量の酢酸 (pKa 4.8) 存在下にMe2CuLiを加えるとMeLi換算で10当量必要としたものの、ほぼ目的物Aのみが得られた。さすがに10当量はまずいだろうということで、フェノール (PKa 10) とメタノール (pKa 15.5) を共存させて付加反応をやってもらった。メタノールは何の効果も無かったが、フェノールではMe2CuLiの量を4当量に抑えることができた。
例えばシクロヘキサノンのしリルエノールエーテルを合成するにはケトンとMe3SiClを共存させておいてLDAを加えると目的物が得られるが、(1-1) と過剰のMe3SiClの共存下にMe2CuLiを加えても目的物A (1-2) は全く得られず、不要なB (1-3) のみが得られた。そこで1当量のBF3•Et2O存在下に反応を行ったところ初めて目的物Aと不要物Bが1:1の比で得られてきた。これはいいぞと思い10当量のBF3•Et2Oを用いたところ、わずかに目的物が増えたのみだった。ここで色々考えたのだが、「窮鼠猫を噛む」の例え通り、酢酸存在下で反応を行うことにした。これはただの勘なのだが、エノレートをquenchする最速の種はプロトンであると思ったことと、院生の頃に化学科の図書館でChemistry & Industryという英国で隔週出版されたHighlightsの中にブロモベンゼンとD2O共存下にBuLiを加えるとD化されたベンゼンが生成するという驚き記事を見た記憶があったからだ。おそらくBuLiからPhBrへの電子移動の方がD2Oへのそれより速いということだろうが、Me2CuLiの反応機構も電子移動が先行するという報告を見たことがあった。神田さんは私の無茶なリクエストに驚いただろうが、一応理屈だけはうまくいくかも知れないという期待はあった。果たして、5当量の酢酸 (pKa 4.8) 存在下にMe2CuLiを加えるとMeLi換算で10当量必要としたものの、ほぼ目的物Aのみが得られた。さすがに10当量はまずいだろうということで、フェノール (PKa 10) とメタノール (pKa 15.5) を共存させて付加反応をやってもらった。メタノールは何の効果も無かったが、フェノールではMe2CuLiの量を4当量に抑えることができた。
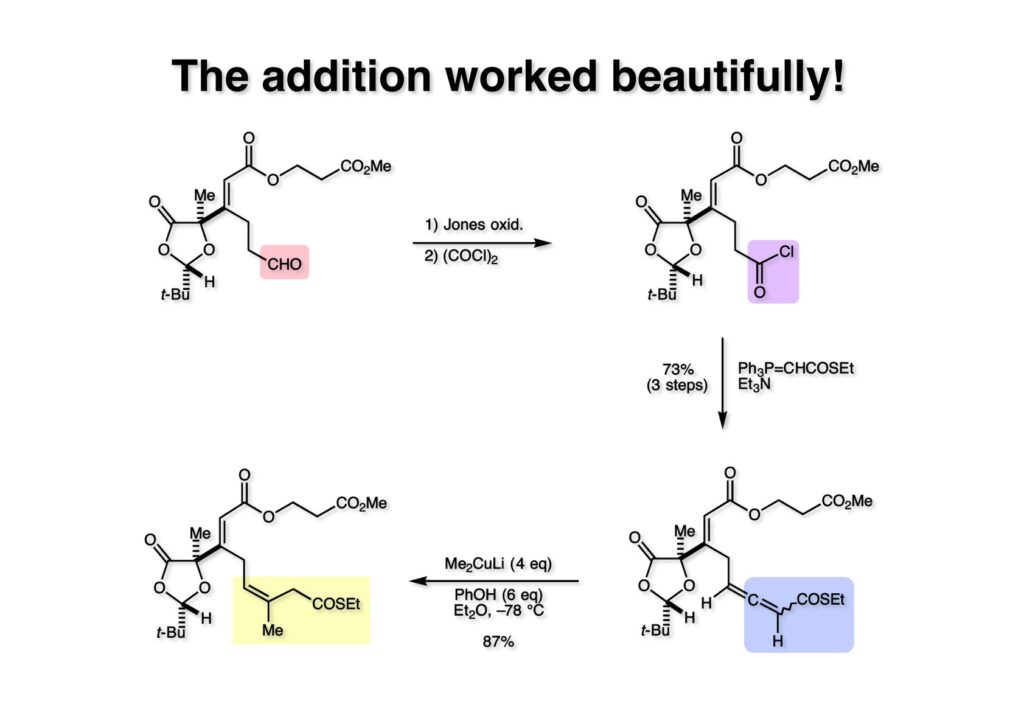 実際に全合成で用いた中間体 (1-1) のアルデヒドをJones酸化してカルボン酸に変換し、さらに塩化オキザリルを用いて酸クロライド (1-2) を得た。これとチオエステルの安定イリド (2-1) の混合物にEt3Nを加えてアレンチオエステルのジアステレオマー混合物 (3-2) を得た。次に、(3-2) と6当量のフェノール存在下でMe2CuLi (4当量) を加えたところ目的物 (3-1) が87%の高収率で得られた。地味ながらも「やった!」という貴重な反応である。
実際に全合成で用いた中間体 (1-1) のアルデヒドをJones酸化してカルボン酸に変換し、さらに塩化オキザリルを用いて酸クロライド (1-2) を得た。これとチオエステルの安定イリド (2-1) の混合物にEt3Nを加えてアレンチオエステルのジアステレオマー混合物 (3-2) を得た。次に、(3-2) と6当量のフェノール存在下でMe2CuLi (4当量) を加えたところ目的物 (3-1) が87%の高収率で得られた。地味ながらも「やった!」という貴重な反応である。
 「反応開発」の項でそのうち記述するが、チオエステルをEt3SiHとPd/Cで反応させるとアルデヒドに変換できる反応を当研究室で開発した。Neothramycin A Me ether (1-3) の全合成でその有用性を検証したが、グルタミン酸由来のチオエステル (1-1) をこの反応条件に付すと、非常に不安定な中間体 (1-2) が良好な収率で得られた。この変換は前代未聞であると自負している。光学活性cyanocycline A (2-3) の全合成ではグルタミン酸誘導体 (2-1) を10 g スケールでアセタール (2-2) に高収率で変換した。Leinamycinの全合成途上でチオエステルを用いているのは勿論その反応を使って有用性を示す意義がある。
「反応開発」の項でそのうち記述するが、チオエステルをEt3SiHとPd/Cで反応させるとアルデヒドに変換できる反応を当研究室で開発した。Neothramycin A Me ether (1-3) の全合成でその有用性を検証したが、グルタミン酸由来のチオエステル (1-1) をこの反応条件に付すと、非常に不安定な中間体 (1-2) が良好な収率で得られた。この変換は前代未聞であると自負している。光学活性cyanocycline A (2-3) の全合成ではグルタミン酸誘導体 (2-1) を10 g スケールでアセタール (2-2) に高収率で変換した。Leinamycinの全合成途上でチオエステルを用いているのは勿論その反応を使って有用性を示す意義がある。
 チオエステルからアルデヒドやケトンへ変換する総説は当時助教授だった徳山さん(現東北大学教授)におんぶに抱っこでAldrichimica Actaに書いてもらった。興味のある方がご覧あれ。
チオエステルからアルデヒドやケトンへ変換する総説は当時助教授だった徳山さん(現東北大学教授)におんぶに抱っこでAldrichimica Actaに書いてもらった。興味のある方がご覧あれ。
 (1-1) をEt3SiH-10% Pd/Cで処理すると円滑に還元が進行してアルデヒド (1-2) が得られた。これを共役アルデヒドにするためにアミン塩基であるDABCOを用いたところ室温で二重結合が移動して目的とするE体アルデヒド (2-2) のみが得られた。E体の生成は速度論的に有利であったが、必ずしも熱力学的に圧倒的に有利ではなく、反応時間を長くするとZ体も徐々に生成してきた。これにてleinamycin (2-1) の水色の部分の合成が完了したことになる。
(1-1) をEt3SiH-10% Pd/Cで処理すると円滑に還元が進行してアルデヒド (1-2) が得られた。これを共役アルデヒドにするためにアミン塩基であるDABCOを用いたところ室温で二重結合が移動して目的とするE体アルデヒド (2-2) のみが得られた。E体の生成は速度論的に有利であったが、必ずしも熱力学的に圧倒的に有利ではなく、反応時間を長くするとZ体も徐々に生成してきた。これにてleinamycin (2-1) の水色の部分の合成が完了したことになる。
 次の課題としては (1-1) のC-8位の立体化学を制御することである。逆合成計画時から不斉アルドール反応を用いるつもりで、Evansのキラル補助基を使ってそこそこの結果を得ていた。ちょうどその頃、日米有機化学セミナーが東京で開催され、私は米国側の一員として参加した。そこでチオエステルのシリルエノールエーテルを使った向山不斉アルドール反応を当時理科大の小林修さんが発表していて、これを用いれば1段階減らすことができると思って神田さんに次ページのような反応をやってほしいと頼んだ。
次の課題としては (1-1) のC-8位の立体化学を制御することである。逆合成計画時から不斉アルドール反応を用いるつもりで、Evansのキラル補助基を使ってそこそこの結果を得ていた。ちょうどその頃、日米有機化学セミナーが東京で開催され、私は米国側の一員として参加した。そこでチオエステルのシリルエノールエーテルを使った向山不斉アルドール反応を当時理科大の小林修さんが発表していて、これを用いれば1段階減らすことができると思って神田さんに次ページのような反応をやってほしいと頼んだ。
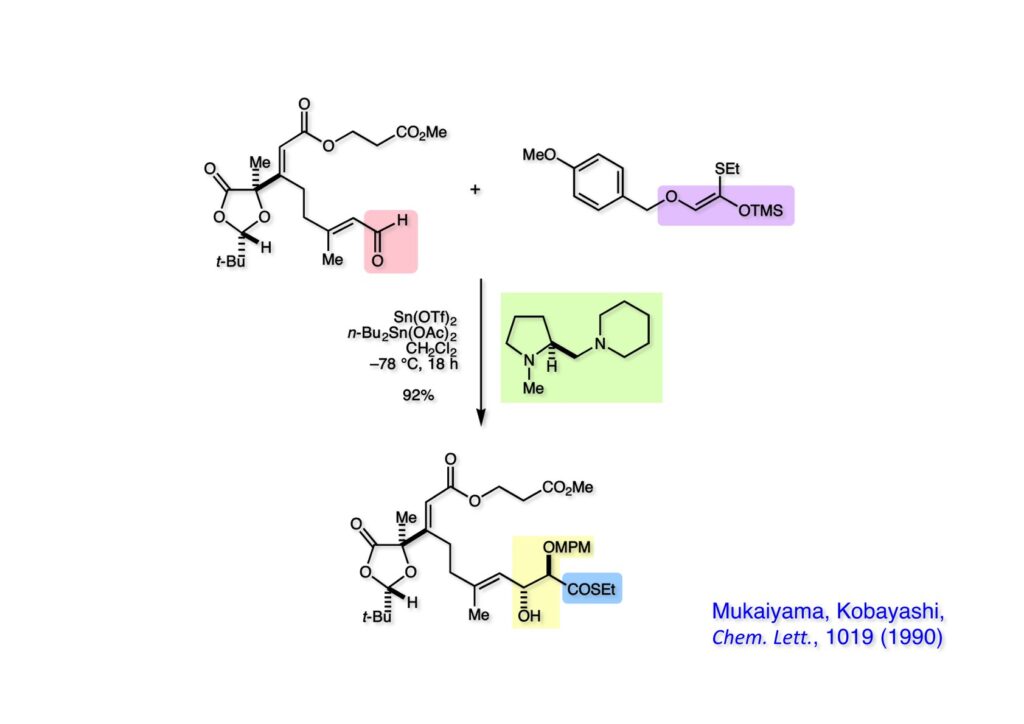 チオエステルのシリルエノールエーテル (1-2) の都合が良いところは、チオエステルから合成する際にE体とZ体 (1-2) の混合物となるが、アルドール反応ではZ体のみが反応することである。アルデヒド (1-1) とシリルエノールエーテル (1-2) の混合物をキラルジアミン (2-1) 存在下で上記の条件に付すと目的とするアルドール付加体 (3-1) が高収率で得られた。-78度で16時間っていう条件がちょっときついが、背に腹は代えられない。
チオエステルのシリルエノールエーテル (1-2) の都合が良いところは、チオエステルから合成する際にE体とZ体 (1-2) の混合物となるが、アルドール反応ではZ体のみが反応することである。アルデヒド (1-1) とシリルエノールエーテル (1-2) の混合物をキラルジアミン (2-1) 存在下で上記の条件に付すと目的とするアルドール付加体 (3-1) が高収率で得られた。-78度で16時間っていう条件がちょっときついが、背に腹は代えられない。
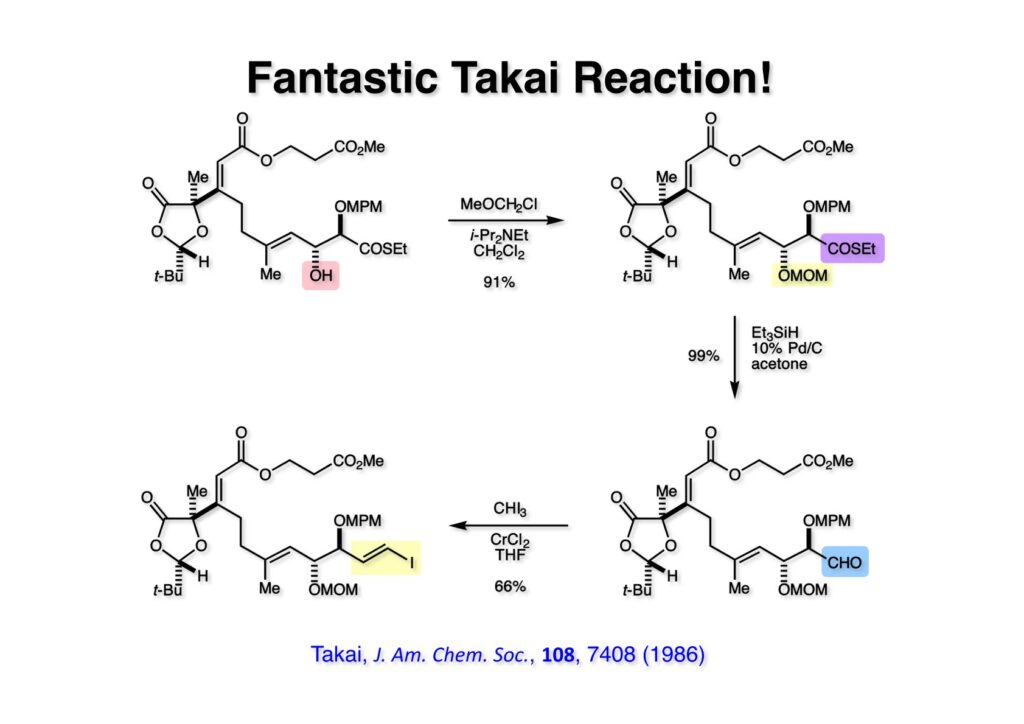 まずアルドール付加体 (1-1) の水酸基をMOM基で保護して (1-2) とし、次に福山還元でチオエステルをアルデヒド (2-2) に99%の収率で変換した。このアルデヒドの増炭に高井反応を用いることは当初からの計画で、実際、穏和な条件でE体のヨウ化ビニル体 (2-1) が主生成物として得られた。高井さん(当時は岡山大)は京大工学部野崎研で檜山為次郎さんの後輩(弟子みたいなもの)で、岸研で檜山さんと同じ部屋で一年過ごした時に時々高井さんの話を檜山さんから聞いていたので親しみが湧いていた。高井反応は広い官能基共存性を持つ有用な反応であるのは間違いない。
まずアルドール付加体 (1-1) の水酸基をMOM基で保護して (1-2) とし、次に福山還元でチオエステルをアルデヒド (2-2) に99%の収率で変換した。このアルデヒドの増炭に高井反応を用いることは当初からの計画で、実際、穏和な条件でE体のヨウ化ビニル体 (2-1) が主生成物として得られた。高井さん(当時は岡山大)は京大工学部野崎研で檜山為次郎さんの後輩(弟子みたいなもの)で、岸研で檜山さんと同じ部屋で一年過ごした時に時々高井さんの話を檜山さんから聞いていたので親しみが湧いていた。高井反応は広い官能基共存性を持つ有用な反応であるのは間違いない。
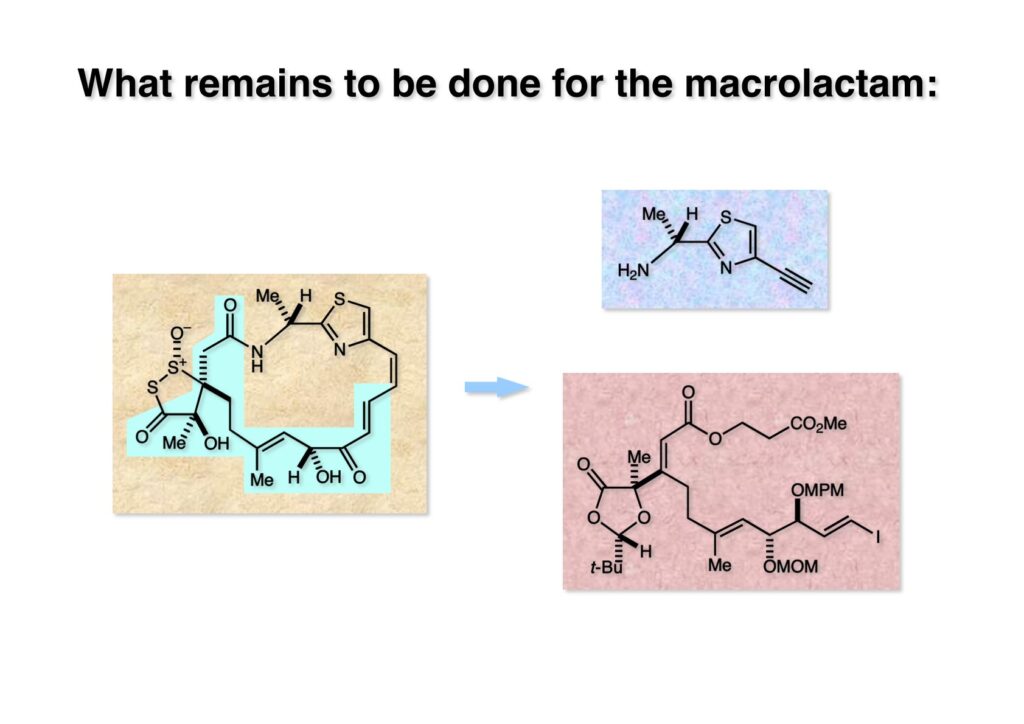 前ページまででleinamycin (2-1) の水色の部分の合成が終わった。次はヨウ化ビニル体 (3-1) からチアゾールアセチレン (1-1) を用いて薗頭カップリングを行い、続いてアセチレンを部分還元してcis-オレフィンに変換すればマクロラクタム化に到達することができる。Chemical Abstractsで4位にアセチレンを有するチアゾールを探したが見つからなかった。これは変だぞ、ということでCAS-ONLINE (SciFinderの前身) の部分構造検索を行ったが見つからなかった。当時はこの部分検索が高くて1件150ドルしたが空振りに終わった(勿体無い)。まあ、見つからないので普通に合成するしかないと次ページのような合成を行った。
前ページまででleinamycin (2-1) の水色の部分の合成が終わった。次はヨウ化ビニル体 (3-1) からチアゾールアセチレン (1-1) を用いて薗頭カップリングを行い、続いてアセチレンを部分還元してcis-オレフィンに変換すればマクロラクタム化に到達することができる。Chemical Abstractsで4位にアセチレンを有するチアゾールを探したが見つからなかった。これは変だぞ、ということでCAS-ONLINE (SciFinderの前身) の部分構造検索を行ったが見つからなかった。当時はこの部分検索が高くて1件150ドルしたが空振りに終わった(勿体無い)。まあ、見つからないので普通に合成するしかないと次ページのような合成を行った。
 幸いなことにChemical Abstractsの検索で安価なL-乳酸 (1-1) から比較的効率よく (1-2) のような光学活性エステル体 (1-2) が得られるということがオーストリアのUhlrich Schmidtによって報告されていた。彼の研究は他の全合成にも使った覚えがあるが、何だったかは思い出せない。(1-2) の水酸基をTBS基で保護してからLiAlH4でエステルを還元してアルコール (1-3) を得た。次にSwern酸化でアルデヒドに変換しCorey-Fuchs法でまずジブロモオレフィン (2-3) を得た。チオエステルの福山還元でアルデヒドを合成したいところだが、エステルを加水分解してチオエステルにしなければならないので断念した。(2-3) を低温下BuLi処理するとアセチレンが生成し、TBAFでTBS基を脱保護することでアセチレン体 (2-2) を得た。次にHN3を使って光延反応を行うと (2-2) はほぼ瞬時に定量的にアジド体 (2-1) に変換することができた。神田さんの博士論文が無いので詳細は不明だた、私の常識を書けば、DPPAを使った光延反応はHN3に比べるとかなり遅く、反応の収率も低めだったと思う。それとHN3を発生させるのは反応に使うトルエン中でNaN3とH2SO4を使っていたと思う。この条件ではNa2SO4が生成し、脱水剤として作用するのでH2Oによる反応阻害が起きにくい。結局4位にアセチレンを有するチアゾールは問題なく合成でき、それまで誰も合成する必要が無かっただけかもしれない。
幸いなことにChemical Abstractsの検索で安価なL-乳酸 (1-1) から比較的効率よく (1-2) のような光学活性エステル体 (1-2) が得られるということがオーストリアのUhlrich Schmidtによって報告されていた。彼の研究は他の全合成にも使った覚えがあるが、何だったかは思い出せない。(1-2) の水酸基をTBS基で保護してからLiAlH4でエステルを還元してアルコール (1-3) を得た。次にSwern酸化でアルデヒドに変換しCorey-Fuchs法でまずジブロモオレフィン (2-3) を得た。チオエステルの福山還元でアルデヒドを合成したいところだが、エステルを加水分解してチオエステルにしなければならないので断念した。(2-3) を低温下BuLi処理するとアセチレンが生成し、TBAFでTBS基を脱保護することでアセチレン体 (2-2) を得た。次にHN3を使って光延反応を行うと (2-2) はほぼ瞬時に定量的にアジド体 (2-1) に変換することができた。神田さんの博士論文が無いので詳細は不明だた、私の常識を書けば、DPPAを使った光延反応はHN3に比べるとかなり遅く、反応の収率も低めだったと思う。それとHN3を発生させるのは反応に使うトルエン中でNaN3とH2SO4を使っていたと思う。この条件ではNa2SO4が生成し、脱水剤として作用するのでH2Oによる反応阻害が起きにくい。結局4位にアセチレンを有するチアゾールは問題なく合成でき、それまで誰も合成する必要が無かっただけかもしれない。
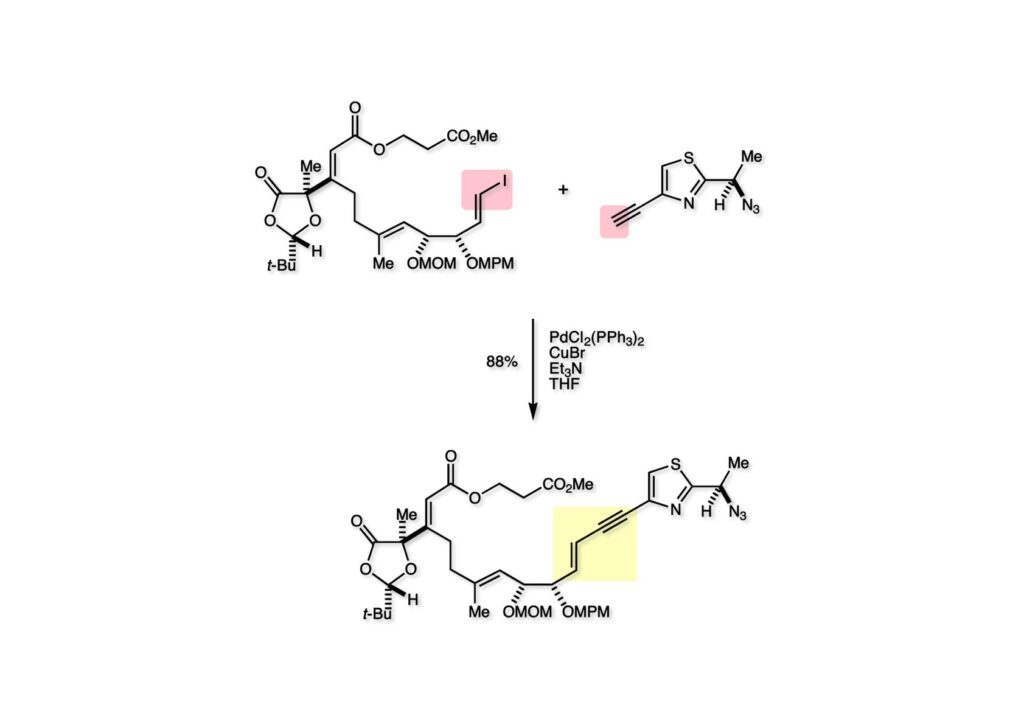 ヨウ化ビニル (1-1) とアセチレン (1-2) の薗頭反応は良好に進行しカップリング体 (2-1) が高収率で得られた。やはり高い官能基共存性のある反応は全合成にとって有難い味方である。
ヨウ化ビニル (1-1) とアセチレン (1-2) の薗頭反応は良好に進行しカップリング体 (2-1) が高収率で得られた。やはり高い官能基共存性のある反応は全合成にとって有難い味方である。
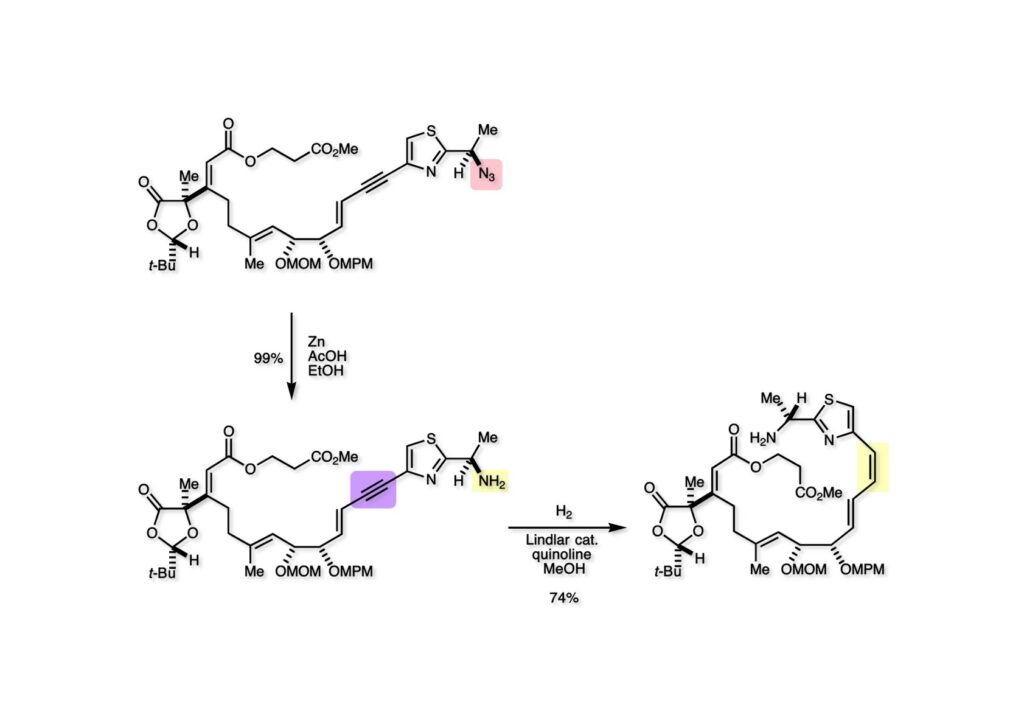 始めはアジドのアミンへの還元とアセチレンの部分還元をLindlar触媒で一挙に行おうとしたが、アセチレンの部分還元が非常に微妙で過剰還元された生成物が生じてきたのでアジドを亜鉛還元してからアセチレンの部分還元をすることにした。部分還元はTLCを何度も展開してモニターしなければならなかったがまずまずの収率でtrans, cis-ジエン (2-2) を得ることができた。一般的に言って、芳香環や二重結合に共役したアセチレンの部分還元は非常に難しくよほど注意深くしないと過剰還元が起きてしまうので気をつけるべきである。
始めはアジドのアミンへの還元とアセチレンの部分還元をLindlar触媒で一挙に行おうとしたが、アセチレンの部分還元が非常に微妙で過剰還元された生成物が生じてきたのでアジドを亜鉛還元してからアセチレンの部分還元をすることにした。部分還元はTLCを何度も展開してモニターしなければならなかったがまずまずの収率でtrans, cis-ジエン (2-2) を得ることができた。一般的に言って、芳香環や二重結合に共役したアセチレンの部分還元は非常に難しくよほど注意深くしないと過剰還元が起きてしまうので気をつけるべきである。
 さて、いよいよマクロラクタム化の出番である。前述のようにここまでの条件検討などはメチルエステル体を用いてやってきた。ところがジオキソラノン環存在下ではメチルエステルの加水分解はできないので、長ったらしいメチルエステルを持つ保護基を使ってきた。いよいよretro-Michael反応でその保護基を落とさなければならない。それには (1-1) をアセトニトリル中室温でDBUを用いて処理した。90分後にはほぼ定量的にカルボン酸 (2-1) が得られた。肝心のマクロラクタム化だが、高希釈条件を使わなくともBOP-Clで60度20分であっという間に終わってしまったのは拍子抜けだった。拍子抜けだった。おそらくsp2炭素が多数あってコンフォメーションがかなり固定され、両末端が近くに存在していたのでは無いかと推察している。
さて、いよいよマクロラクタム化の出番である。前述のようにここまでの条件検討などはメチルエステル体を用いてやってきた。ところがジオキソラノン環存在下ではメチルエステルの加水分解はできないので、長ったらしいメチルエステルを持つ保護基を使ってきた。いよいよretro-Michael反応でその保護基を落とさなければならない。それには (1-1) をアセトニトリル中室温でDBUを用いて処理した。90分後にはほぼ定量的にカルボン酸 (2-1) が得られた。肝心のマクロラクタム化だが、高希釈条件を使わなくともBOP-Clで60度20分であっという間に終わってしまったのは拍子抜けだった。拍子抜けだった。おそらくsp2炭素が多数あってコンフォメーションがかなり固定され、両末端が近くに存在していたのでは無いかと推察している。
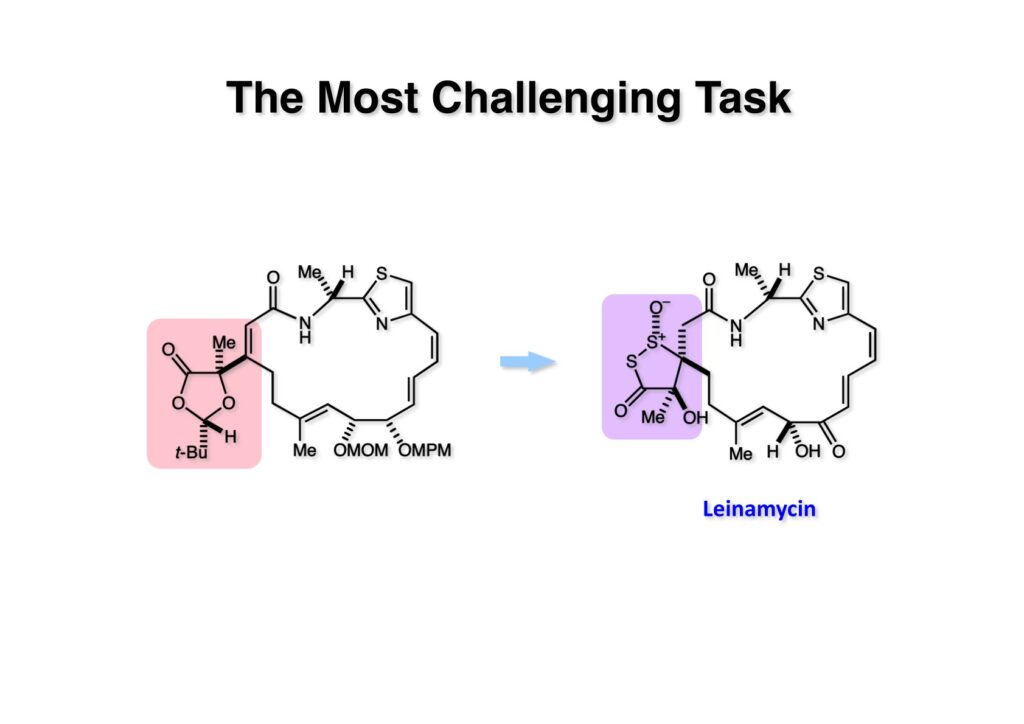 マクロラクタム環は無事に構築することができたが、(1-1) からジチオラノン骨格を持つleinamycin (1-2) に変換するのは未知のロードで、気分的にはやっと道半ばに達したというところだった。
マクロラクタム環は無事に構築することができたが、(1-1) からジチオラノン骨格を持つleinamycin (1-2) に変換するのは未知のロードで、気分的にはやっと道半ばに達したというところだった。
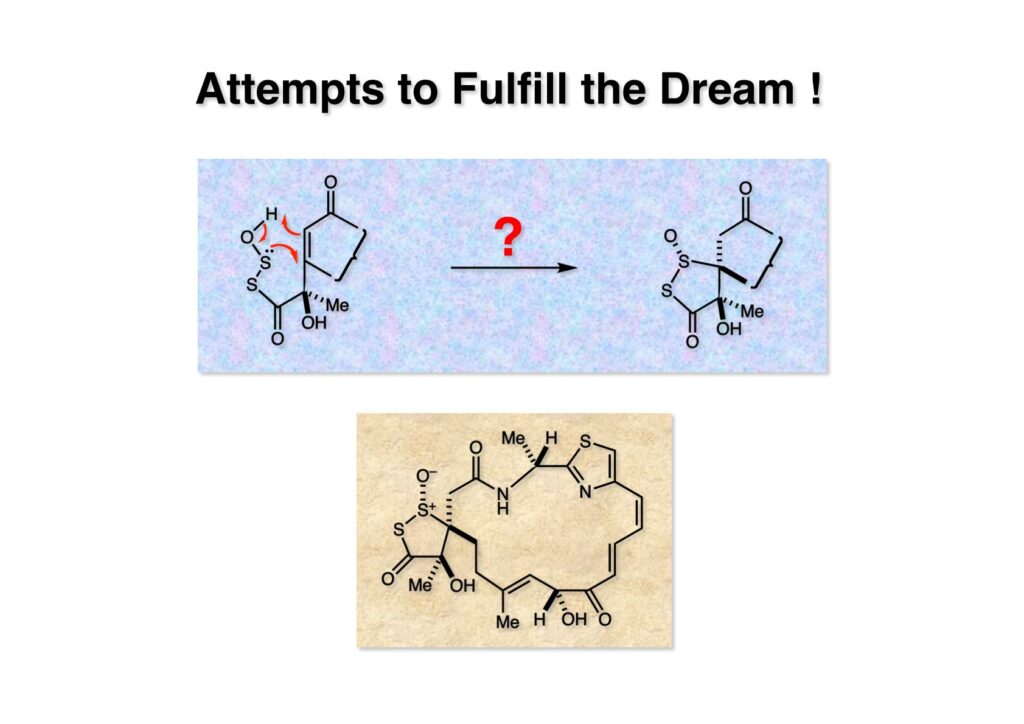 まず最初にやろうと思ったのは (1-1) のような未知の化学種を生成して一挙に目的とする環 (1-2) を構築するというアイデアだ。研究を進めるにあたり、これが成功したら素晴らしいだろうな、というようなアイデアが浮かんだら、まずそれを実行してみるようでなければ面白くない。石橋を叩きながら地を這うようにして目的地点に恐る恐る辿り着くようでは新しい反応や変換方法は見つからないと思う。全合成においては特にそう言えるだろう。反応開発では目的とする反応がうまく進行しなくても、予想外の面白い反応が見つかることは勿論あるが。ここではスルホキサイド脱離の逆反応みたいなのが起これば良いなという考えでモデル実験を行なってみた。
まず最初にやろうと思ったのは (1-1) のような未知の化学種を生成して一挙に目的とする環 (1-2) を構築するというアイデアだ。研究を進めるにあたり、これが成功したら素晴らしいだろうな、というようなアイデアが浮かんだら、まずそれを実行してみるようでなければ面白くない。石橋を叩きながら地を這うようにして目的地点に恐る恐る辿り着くようでは新しい反応や変換方法は見つからないと思う。全合成においては特にそう言えるだろう。反応開発では目的とする反応がうまく進行しなくても、予想外の面白い反応が見つかることは勿論あるが。ここではスルホキサイド脱離の逆反応みたいなのが起これば良いなという考えでモデル実験を行なってみた。
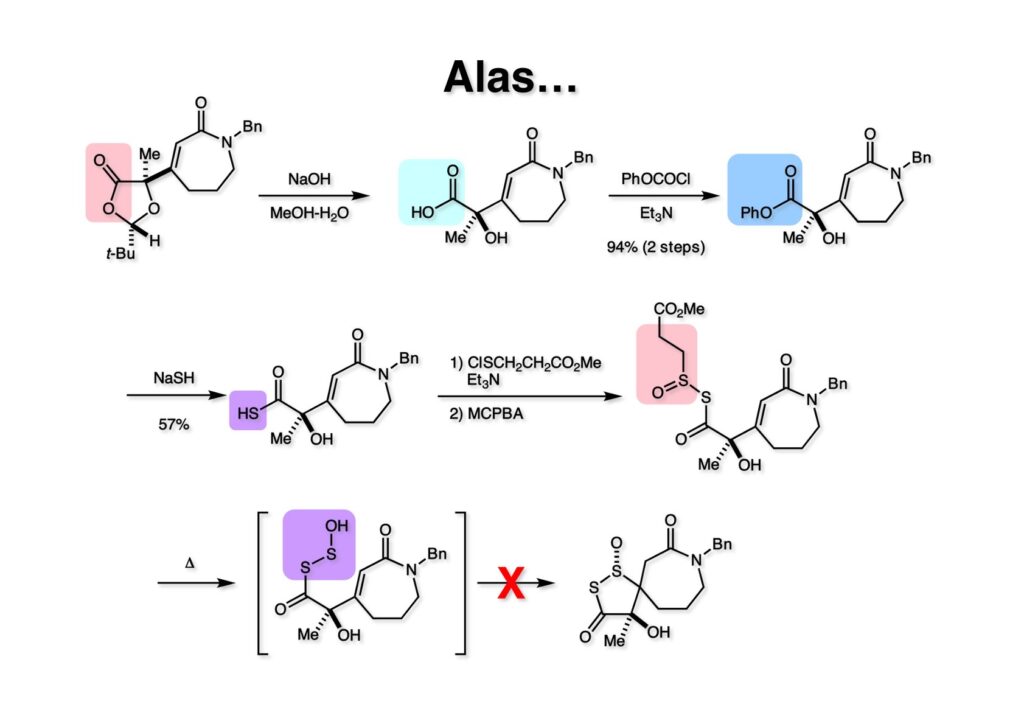 モデル化合物 (1-1) は合成中間体のアルデヒドをPhCH2NH2でreductive aminationすることで容易に得られる。ジオキソラノン (1-1) をアルカリ加水分解すると (1-2) が得られる。これを PhOCOClとEt3Nで加熱するとフェニルエステル (1-3) が得られるが反応機構的には少し面白い。中間体は5員環の混合酸無水物でこれにPhO-がアタックすると (1-3) が生成する。勿論フェニルエステルは反応性が高いのでNaSHで処理するとチオカルボン酸 (2-1) が得られる。チオカルボン酸に塩化スルフェニル (2-2) を反応させるとジスルフィド類縁体が生成し、さらに1当量のMCPBAで酸化すると (2-3) が得られた。(2-3) を加熱して脱離を行えば (3-1) が生成して環化反応により (3-2) が得られることを期待したのだが、残念ながら目的物は痕跡も得られなかった。このアイデアをやらずして先に進ことはできないと思っていたので、より実現性の高そうなアイデアを実行に移すことにした。
モデル化合物 (1-1) は合成中間体のアルデヒドをPhCH2NH2でreductive aminationすることで容易に得られる。ジオキソラノン (1-1) をアルカリ加水分解すると (1-2) が得られる。これを PhOCOClとEt3Nで加熱するとフェニルエステル (1-3) が得られるが反応機構的には少し面白い。中間体は5員環の混合酸無水物でこれにPhO-がアタックすると (1-3) が生成する。勿論フェニルエステルは反応性が高いのでNaSHで処理するとチオカルボン酸 (2-1) が得られる。チオカルボン酸に塩化スルフェニル (2-2) を反応させるとジスルフィド類縁体が生成し、さらに1当量のMCPBAで酸化すると (2-3) が得られた。(2-3) を加熱して脱離を行えば (3-1) が生成して環化反応により (3-2) が得られることを期待したのだが、残念ながら目的物は痕跡も得られなかった。このアイデアをやらずして先に進ことはできないと思っていたので、より実現性の高そうなアイデアを実行に移すことにした。
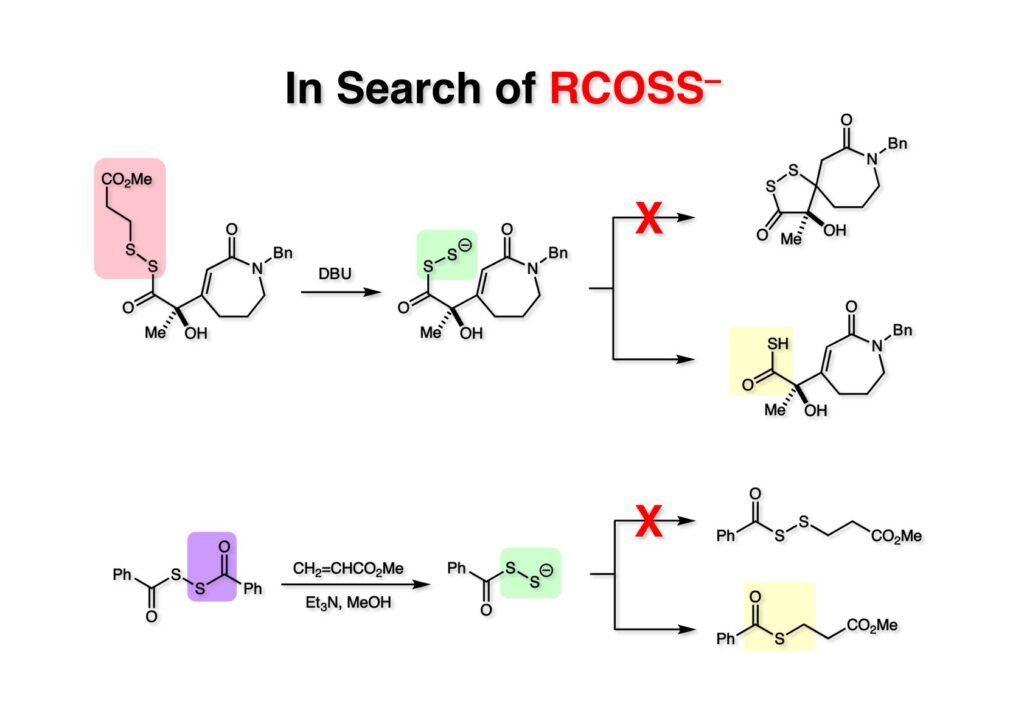 次に可能性を試そうと思った反応はRCOSS-という、これも又使われたことも無いような種がSを放出してRCOS-になる前にα,β-不飽和ラクタムにMichael付加するかどうかだ。(1-1) をDBUを使ってretro-Michael反応で (1-2) を生成して共役付加するかどうかを見てみたが、残念ながら (1-3) は得られず、チオカルボン酸 (2-1) が得られるのみだった。一体全体RCOSS- (3-2) は生成するのかどうかを大過剰のアクリル酸メチルで捕捉できるかどうかを確かめるため、メタノール中で (3-1) をEt3N存在下で加メタノール分解してみた。これも残念ながら (3-3) は得られずに (4-1) が生成しただけだった。おそらく硫黄の脱離反応は非常に速いものと推測した。
次に可能性を試そうと思った反応はRCOSS-という、これも又使われたことも無いような種がSを放出してRCOS-になる前にα,β-不飽和ラクタムにMichael付加するかどうかだ。(1-1) をDBUを使ってretro-Michael反応で (1-2) を生成して共役付加するかどうかを見てみたが、残念ながら (1-3) は得られず、チオカルボン酸 (2-1) が得られるのみだった。一体全体RCOSS- (3-2) は生成するのかどうかを大過剰のアクリル酸メチルで捕捉できるかどうかを確かめるため、メタノール中で (3-1) をEt3N存在下で加メタノール分解してみた。これも残念ながら (3-3) は得られずに (4-1) が生成しただけだった。おそらく硫黄の脱離反応は非常に速いものと推測した。
 そこでダメ元でチオールが共役付加するかどうかを見てみたが、(1-1) も (2-1) も分子間Michael付加は起きなかった。この時点では立体化学がどうのこうのというより、付加が起きるかどうかだけに関心があった。
そこでダメ元でチオールが共役付加するかどうかを見てみたが、(1-1) も (2-1) も分子間Michael付加は起きなかった。この時点では立体化学がどうのこうのというより、付加が起きるかどうかだけに関心があった。
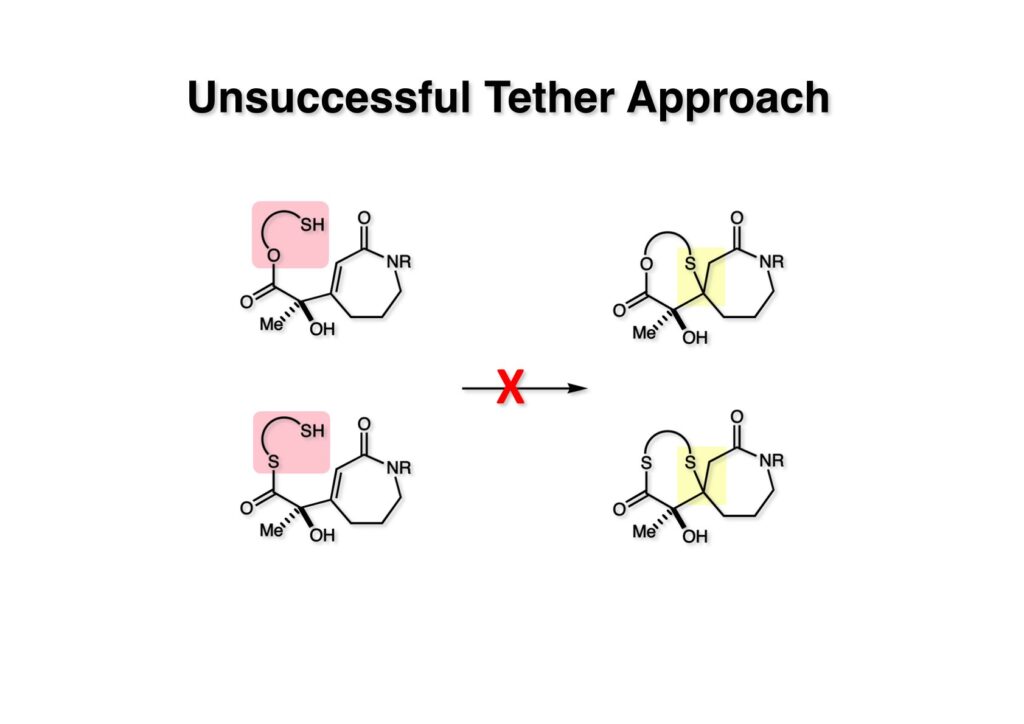 チオレートの分子内共役付加もやってみたが、(1-1) や (2-1) の安定な紐 (tether) がCH2CH2だったので7員環形成という不利な点も重なって期待した結果は得られなかった。
チオレートの分子内共役付加もやってみたが、(1-1) や (2-1) の安定な紐 (tether) がCH2CH2だったので7員環形成という不利な点も重なって期待した結果は得られなかった。
 分子内共役付加の最後の一手は5員環形成である。いくら何でも5員環が出来ない筈はないという期待感でモデル化合物 (1-1) を使って試してみることにした。メチルケトンを作るつもりでジオキソラノンにMeLiを加えたら付加体 (1-2) が分解せずに得られてきた。しめしめとSOCl2-Pyで脱水を試みたところ期待通りエノールエーテル (1-3) が得られた。次に含水アセトニトリル中でNBSを加えたところブロモヒドリン (2-3) が得られた。(2-3) を強塩基でもあるLi2Sで処理したところアセタールが分解して生じたブロモケトンがLi2Sと反応してチオレート (2-2) となり、直ちに分子内共役付加が進行してスピロ体 (2-1) のジアステレオマー混合物が得られた。勿論、メチレン基が余分に存在しているので除去する必要があるが、このメチレン基の除去には2つアイデアがあった。一つはシリルエノールエーテルに誘導してから何とか酸化的に切断する(例えばジオール化)方法である。これよりは魅力的なアイデアはメチレン基のところをニトロソ化してオキシムにし、BeckmannフラグメンテーションでC-C結合を切断することで、そのためのモデル実験を次ページに示す。
分子内共役付加の最後の一手は5員環形成である。いくら何でも5員環が出来ない筈はないという期待感でモデル化合物 (1-1) を使って試してみることにした。メチルケトンを作るつもりでジオキソラノンにMeLiを加えたら付加体 (1-2) が分解せずに得られてきた。しめしめとSOCl2-Pyで脱水を試みたところ期待通りエノールエーテル (1-3) が得られた。次に含水アセトニトリル中でNBSを加えたところブロモヒドリン (2-3) が得られた。(2-3) を強塩基でもあるLi2Sで処理したところアセタールが分解して生じたブロモケトンがLi2Sと反応してチオレート (2-2) となり、直ちに分子内共役付加が進行してスピロ体 (2-1) のジアステレオマー混合物が得られた。勿論、メチレン基が余分に存在しているので除去する必要があるが、このメチレン基の除去には2つアイデアがあった。一つはシリルエノールエーテルに誘導してから何とか酸化的に切断する(例えばジオール化)方法である。これよりは魅力的なアイデアはメチレン基のところをニトロソ化してオキシムにし、BeckmannフラグメンテーションでC-C結合を切断することで、そのためのモデル実験を次ページに示す。
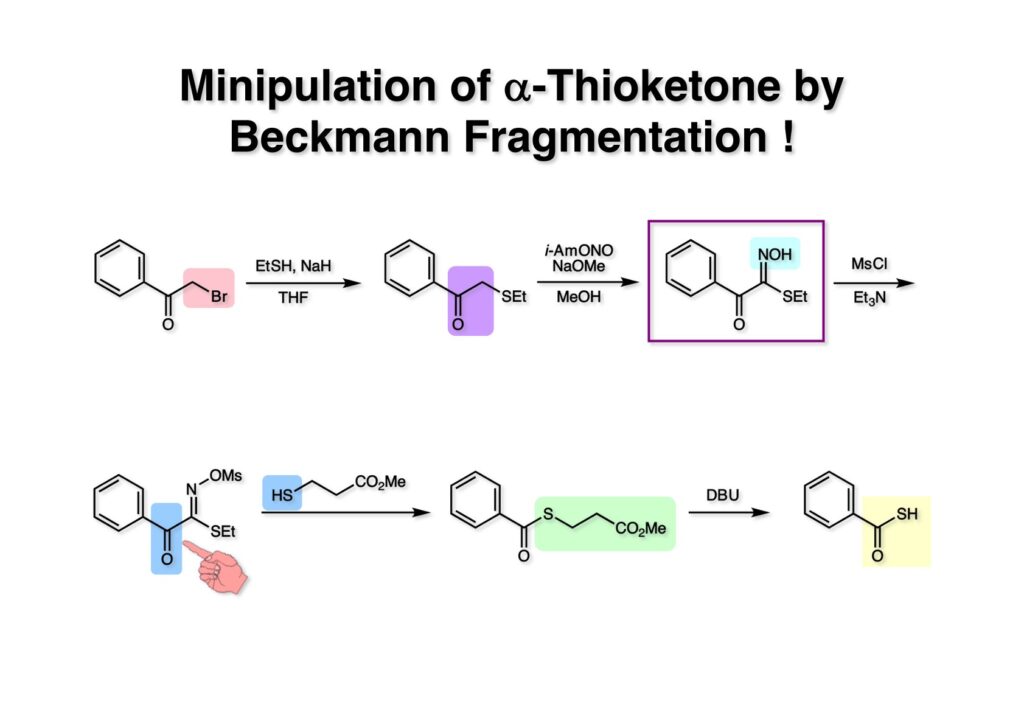 市販のフェナシルブロマイド (1-1) をEtSH-NaHの条件でスルフィド (1-2) に変換した。次にケトンのα位をオキシム化する常法であるi-AmONO-NaOMe/MeOHという条件に (1-2) を付したところ期待通りオキシム (1-3) が得られた。Beckmann fragmentationを円滑に進めるためにオキシムの水酸基をMsClでメシル化して (2-1) に変換した。これを単離することなくHSCH2CH2CO2Meと反応させたところ、期待通りC-C結合の開裂が進行してチオエステル (2-2) が得られた。(2-2) をDBUで処理するとretro-Michael反応が進行しチオカルボン酸 (2-3) が得られた。このモデル実験の成功によりleinamycin全合成への道筋が大筋で決まったと言える。
市販のフェナシルブロマイド (1-1) をEtSH-NaHの条件でスルフィド (1-2) に変換した。次にケトンのα位をオキシム化する常法であるi-AmONO-NaOMe/MeOHという条件に (1-2) を付したところ期待通りオキシム (1-3) が得られた。Beckmann fragmentationを円滑に進めるためにオキシムの水酸基をMsClでメシル化して (2-1) に変換した。これを単離することなくHSCH2CH2CO2Meと反応させたところ、期待通りC-C結合の開裂が進行してチオエステル (2-2) が得られた。(2-2) をDBUで処理するとretro-Michael反応が進行しチオカルボン酸 (2-3) が得られた。このモデル実験の成功によりleinamycin全合成への道筋が大筋で決まったと言える。
 Ethyl vinyl etherの1位の脱プロトン化はt-BuLiを使うのが一般的だが、もう市販されていないようなことを聞いたので今後はどうなるのだろう。私の研究室ではt-BuLiはほとんどない使ったことがないので、おそらくs-BuLi-TMEDAかn-BuLi-t-BuOKを使う1970年代の方法で脱プロトン化したものと思う。これをケトンに付加して得たモデル化合物 (1-1) を含水アセトニトリル中NBSでブロモ化するとブロモケトン (1-2) が得られる。これをTFH中Et3N存在下で硫化水素を吹き込んだところ望む5員環化合物が4:1の比で得られた。NOE実験により主生成物が欲しいもの (1-3) であることが判明した。立体選択性の理由は不明である。このケトン (1-3) をオキシム (2-1) に変換し、その水酸基を2,6-dimethylbenzoate (2-3) として活性化した。メシレートでは反応性が強すぎて扱いにくいのが理由である。2,6-dimethylbenzoateはカルボニル基が立体障害により保護されているので求核剤は簡単に攻撃できない。次に (2-3) を過剰のEtSNaと反応させるとBeckmann fragmentationが進行し、さらにthiocyanide (3-1) もチオールに変換された (3-2) が得られた。(3-2) のチオエステルは反応性が大きいのでNaSHを加えると容易にチオカルボン酸に変換された。これを精製することなくヨウ素で酸化すると望むジチオラノン (3-1) が得られた。このモデル実験によってlenieramycinの全合成の完成に大きく近づいたと言える。
Ethyl vinyl etherの1位の脱プロトン化はt-BuLiを使うのが一般的だが、もう市販されていないようなことを聞いたので今後はどうなるのだろう。私の研究室ではt-BuLiはほとんどない使ったことがないので、おそらくs-BuLi-TMEDAかn-BuLi-t-BuOKを使う1970年代の方法で脱プロトン化したものと思う。これをケトンに付加して得たモデル化合物 (1-1) を含水アセトニトリル中NBSでブロモ化するとブロモケトン (1-2) が得られる。これをTFH中Et3N存在下で硫化水素を吹き込んだところ望む5員環化合物が4:1の比で得られた。NOE実験により主生成物が欲しいもの (1-3) であることが判明した。立体選択性の理由は不明である。このケトン (1-3) をオキシム (2-1) に変換し、その水酸基を2,6-dimethylbenzoate (2-3) として活性化した。メシレートでは反応性が強すぎて扱いにくいのが理由である。2,6-dimethylbenzoateはカルボニル基が立体障害により保護されているので求核剤は簡単に攻撃できない。次に (2-3) を過剰のEtSNaと反応させるとBeckmann fragmentationが進行し、さらにthiocyanide (3-1) もチオールに変換された (3-2) が得られた。(3-2) のチオエステルは反応性が大きいのでNaSHを加えると容易にチオカルボン酸に変換された。これを精製することなくヨウ素で酸化すると望むジチオラノン (3-1) が得られた。このモデル実験によってlenieramycinの全合成の完成に大きく近づいたと言える。
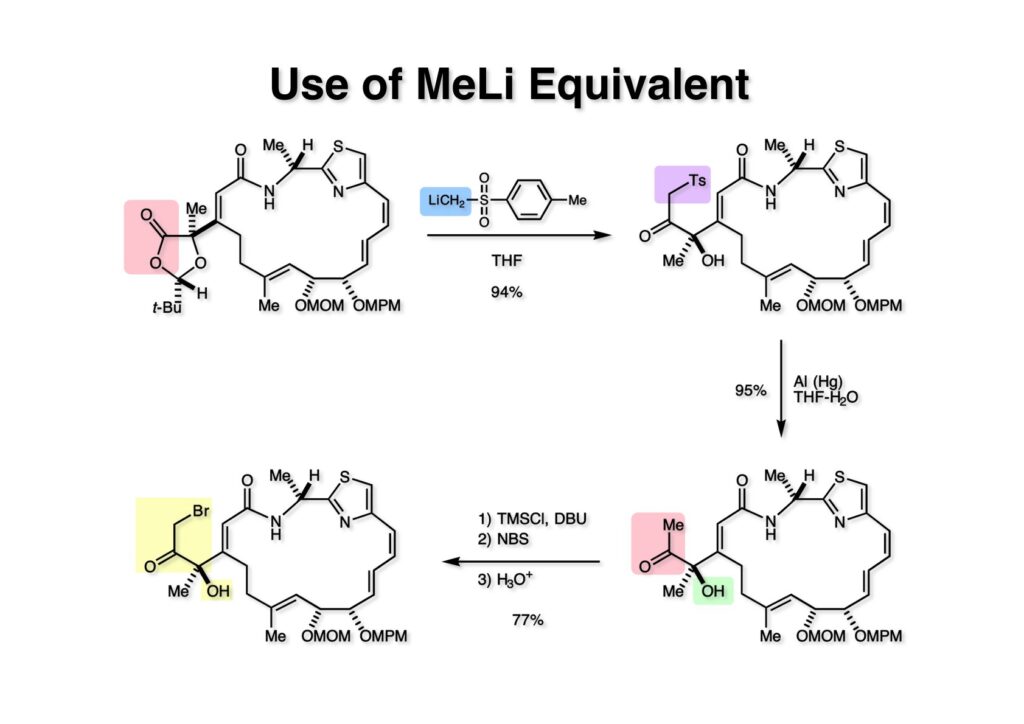 モデル実験がうまくいったので本番に適用しようと中間体 (1-1) に低温下でMeLiを加えたところ、無視できないほど副生成物が出来てしまい改良が必要となった。酢酸エチルのアニオンの付加は非常に綺麗に進行したので、同じくらいのpKaを持つmethyl p-tolyl sulfone (pKa: 25くらい)にBuLiを加えてアニオンにし、それを (1-1) に加えたところ首尾よく目的物 (1-2) が高収率で得られた。このようなトシル基はアルミニウムアマルガムで簡単に除去が可能でメチルケトン (2-2) が高収率で得られた。Al (Hg) の製法は簡単で、アルミホイルをハサミで小さく切って、HgCl2の水溶液中でしばらく攪拌すれば出来上がりである。メチルケトン (2-2) をブロモケトン (2-1) に変換するにはできるだけ穏和な条件を必要とする。まず、TMSClとDBUで加熱するとシリルエノールエーテルと水酸基がシリル化された化合物ができる。これにNBSを作用させるとブロモケトンが精製し、さらに希塩酸を用いてシリルエーテルを除去すれば (2-1) が得られる。
モデル実験がうまくいったので本番に適用しようと中間体 (1-1) に低温下でMeLiを加えたところ、無視できないほど副生成物が出来てしまい改良が必要となった。酢酸エチルのアニオンの付加は非常に綺麗に進行したので、同じくらいのpKaを持つmethyl p-tolyl sulfone (pKa: 25くらい)にBuLiを加えてアニオンにし、それを (1-1) に加えたところ首尾よく目的物 (1-2) が高収率で得られた。このようなトシル基はアルミニウムアマルガムで簡単に除去が可能でメチルケトン (2-2) が高収率で得られた。Al (Hg) の製法は簡単で、アルミホイルをハサミで小さく切って、HgCl2の水溶液中でしばらく攪拌すれば出来上がりである。メチルケトン (2-2) をブロモケトン (2-1) に変換するにはできるだけ穏和な条件を必要とする。まず、TMSClとDBUで加熱するとシリルエノールエーテルと水酸基がシリル化された化合物ができる。これにNBSを作用させるとブロモケトンが精製し、さらに希塩酸を用いてシリルエーテルを除去すれば (2-1) が得られる。
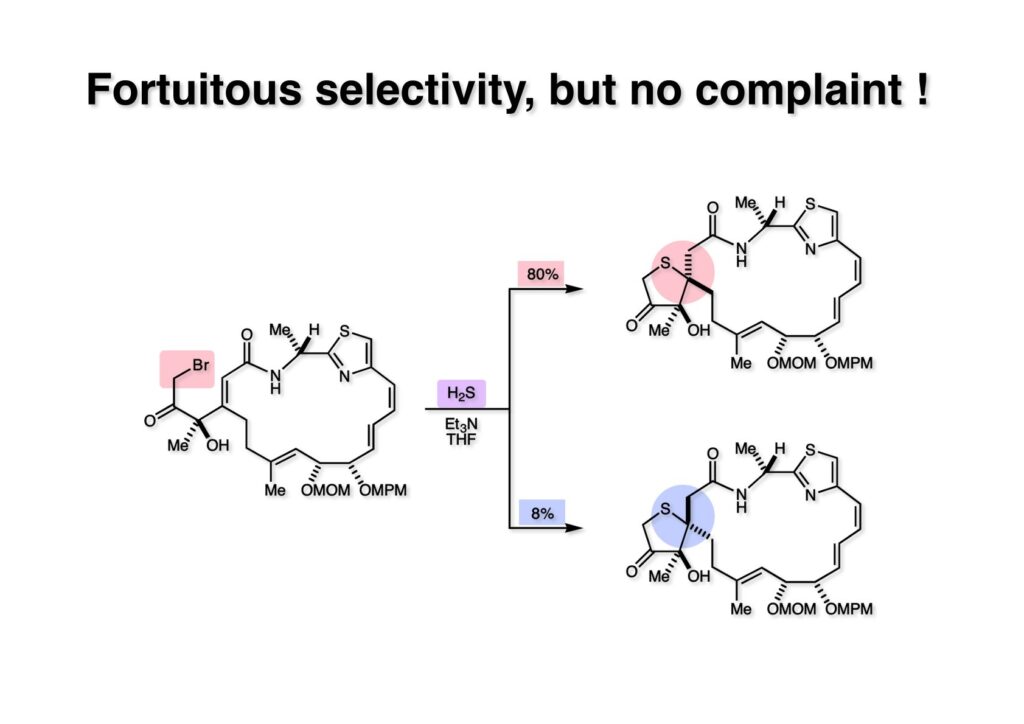 さて、いよいよ運命を左右する反応である。ブロモケトン (1-1) とEt3NのTHF溶液に硫化水素を吹き込んだところ10:1の比でスピロ体の混合物が得られた。主生成物が目的物でなかったら目も当てられないところだが、私の日頃の善行のお陰かNOE実験で主生成物 (80%) が目的物であると判明した。あれこれ苦闘を重ねなくて済んだわけで正直ホッとした。
さて、いよいよ運命を左右する反応である。ブロモケトン (1-1) とEt3NのTHF溶液に硫化水素を吹き込んだところ10:1の比でスピロ体の混合物が得られた。主生成物が目的物でなかったら目も当てられないところだが、私の日頃の善行のお陰かNOE実験で主生成物 (80%) が目的物であると判明した。あれこれ苦闘を重ねなくて済んだわけで正直ホッとした。
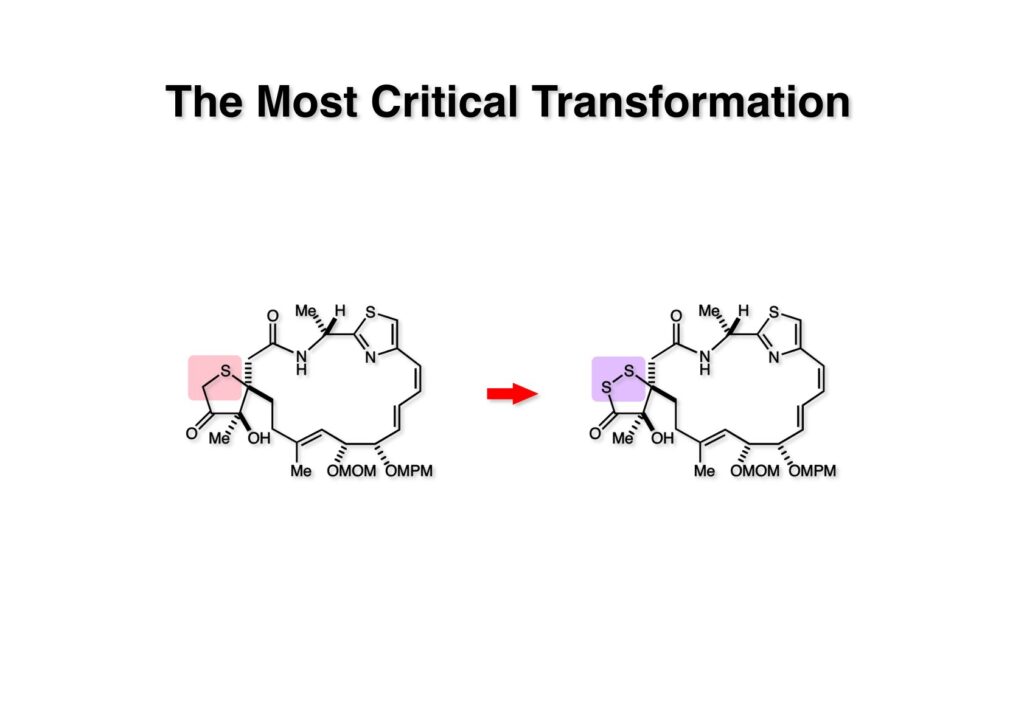 次にやるべきことは (1-1) から(1-2) への変換で、炭素を硫黄に変えるという、まあ錬金術みたいなもので、モデル実験からは確実に実現できるはずの変換であった。
次にやるべきことは (1-1) から(1-2) への変換で、炭素を硫黄に変えるという、まあ錬金術みたいなもので、モデル実験からは確実に実現できるはずの変換であった。
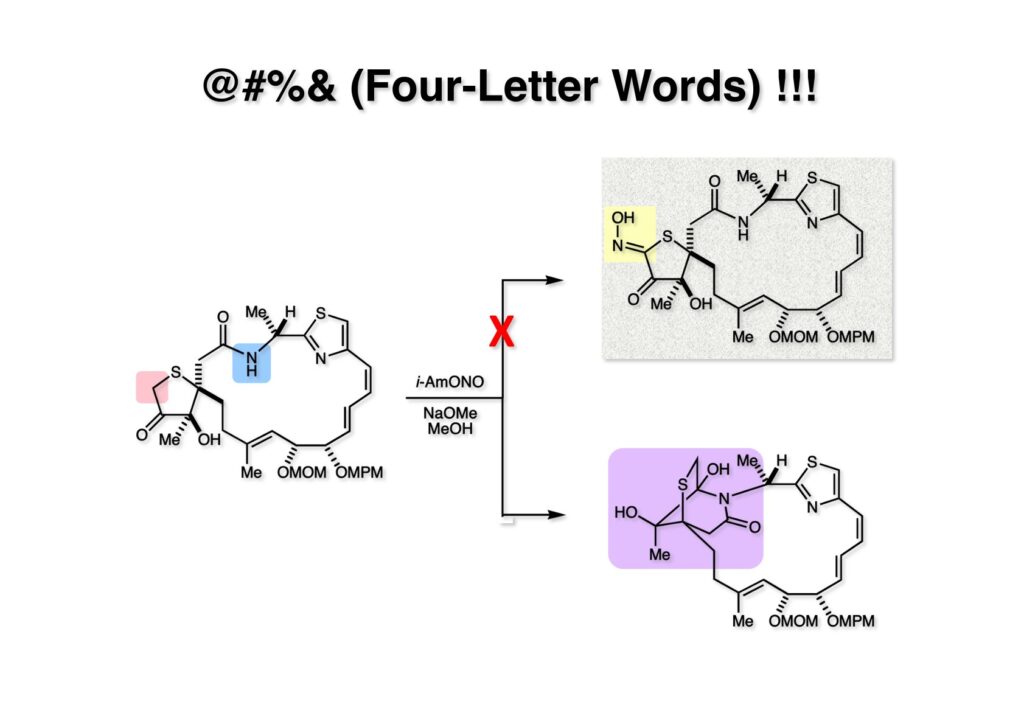 ところが、(2-1) を常法でオキシム (1-1) に変換しようとしたところ、全く目的物 (1-1) が得られなかった!! この条件下でラクタムのNHが脱プロトン化されてケトンに付加した (3-1) になってしまっていた。もうケトンが無いのでオキシムができるわけが無い。(1-1) の段階でBoc基かMOM基で保護するという手が有ることは有るが、保護と脱保護でステップ数が増えてしまう。
ところが、(2-1) を常法でオキシム (1-1) に変換しようとしたところ、全く目的物 (1-1) が得られなかった!! この条件下でラクタムのNHが脱プロトン化されてケトンに付加した (3-1) になってしまっていた。もうケトンが無いのでオキシムができるわけが無い。(1-1) の段階でBoc基かMOM基で保護するという手が有ることは有るが、保護と脱保護でステップ数が増えてしまう。
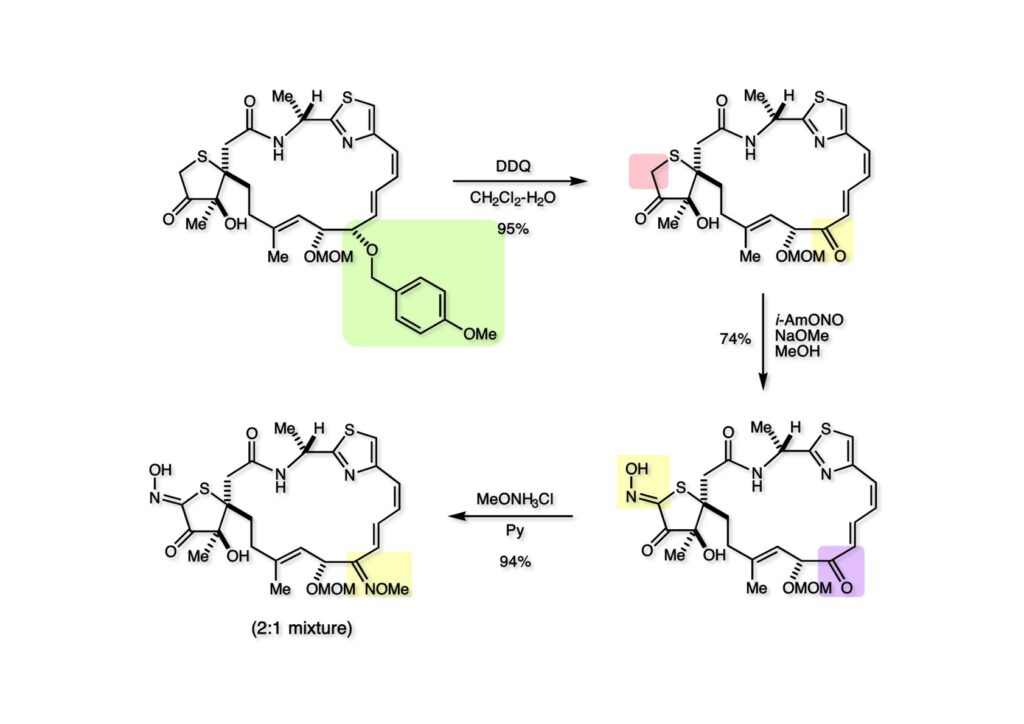 Fieserのデッカい分子模型を眺めながら、何か良い手はないものかと考えた結果、ジエンの部分をジエノンに変換すれば平面性が増加してマクロラクタムの歪みが増すのではないかと思った。(1-1) のPMB基をDDQ酸化で除去しようとしたところジエノン (1-2) がかなり生成していたのでDDQを追加してジエノン (1-2) として単離した。これをオキシム化の条件に付したところ、私の理屈が合っているかどうかは自信が無いものの、見事にオキシム (2-2) が得られてきた。このジエノンにはチオールが付加してしまうので反応性を抑えるためにオキシムエーテル (2-1) に変換しておいた。
Fieserのデッカい分子模型を眺めながら、何か良い手はないものかと考えた結果、ジエンの部分をジエノンに変換すれば平面性が増加してマクロラクタムの歪みが増すのではないかと思った。(1-1) のPMB基をDDQ酸化で除去しようとしたところジエノン (1-2) がかなり生成していたのでDDQを追加してジエノン (1-2) として単離した。これをオキシム化の条件に付したところ、私の理屈が合っているかどうかは自信が無いものの、見事にオキシム (2-2) が得られてきた。このジエノンにはチオールが付加してしまうので反応性を抑えるためにオキシムエーテル (2-1) に変換しておいた。
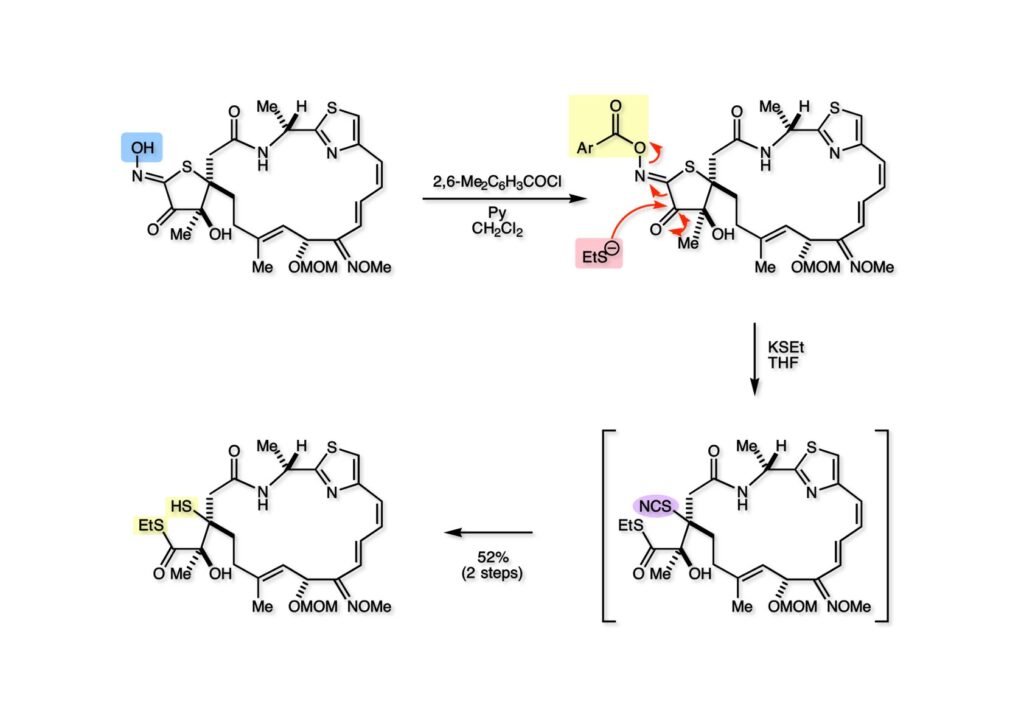 まずオキシム (1-1) の水酸基を2,6-dimethylbenzoate (1-2) に変換し、これに50当量のKSEtを加えてチオエステル (2-1) を得た。実はBeckmann fragmentation自体は1当量のEtSH-Et3Nでも進行するが、生成した (2-2) のthiocyanideに三級水酸基が付加した環状体が出来てしまい、これがEtSHでは開裂しないという面倒な副反応が起きてしまったので、大過剰のEtSKを使って望まない環化が起きる前にthiocyanideをチオールに変換する操作をしたのだ。
まずオキシム (1-1) の水酸基を2,6-dimethylbenzoate (1-2) に変換し、これに50当量のKSEtを加えてチオエステル (2-1) を得た。実はBeckmann fragmentation自体は1当量のEtSH-Et3Nでも進行するが、生成した (2-2) のthiocyanideに三級水酸基が付加した環状体が出来てしまい、これがEtSHでは開裂しないという面倒な副反応が起きてしまったので、大過剰のEtSKを使って望まない環化が起きる前にthiocyanideをチオールに変換する操作をしたのだ。
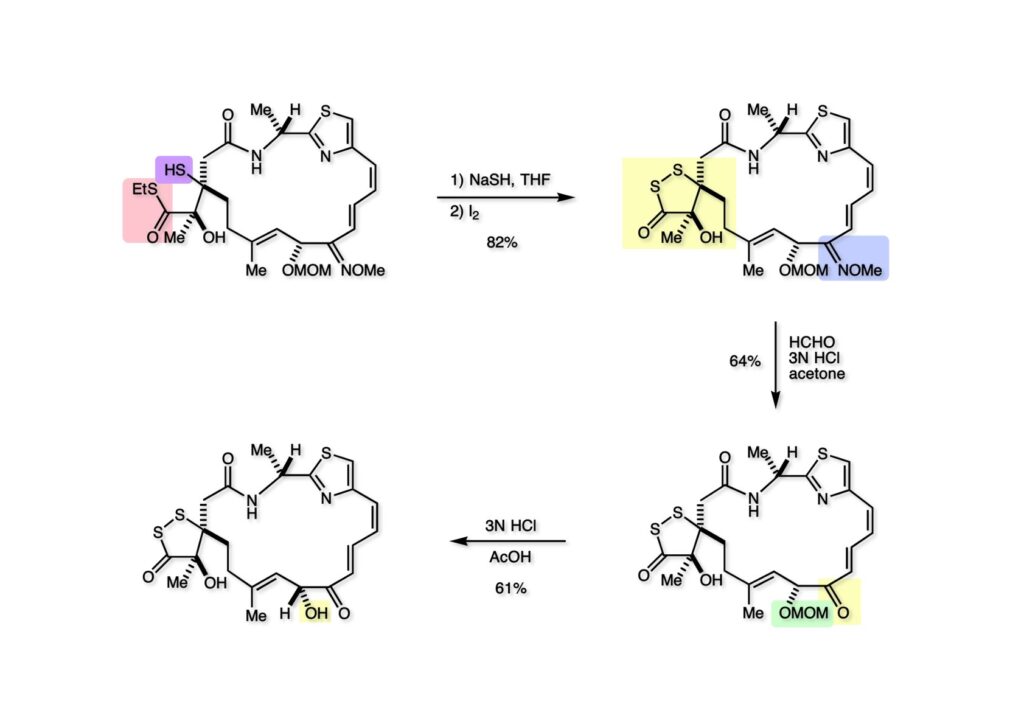 (1-1).をNaSHでチオカルボン酸に変換し、ヨウ素で酸化して無事にdithiolanone (1-2) を得た。次はオキシムエーテル (1-2) をアセトン中でホルムアルデヒドと塩酸で処理することによりオキシムトランスファーを行ってケトン (2-2) が得られた。さらに酢酸中で塩酸を使って注意深くMOM基を除去することで (2-1) を得た。
(1-1).をNaSHでチオカルボン酸に変換し、ヨウ素で酸化して無事にdithiolanone (1-2) を得た。次はオキシムエーテル (1-2) をアセトン中でホルムアルデヒドと塩酸で処理することによりオキシムトランスファーを行ってケトン (2-2) が得られた。さらに酢酸中で塩酸を使って注意深くMOM基を除去することで (2-1) を得た。
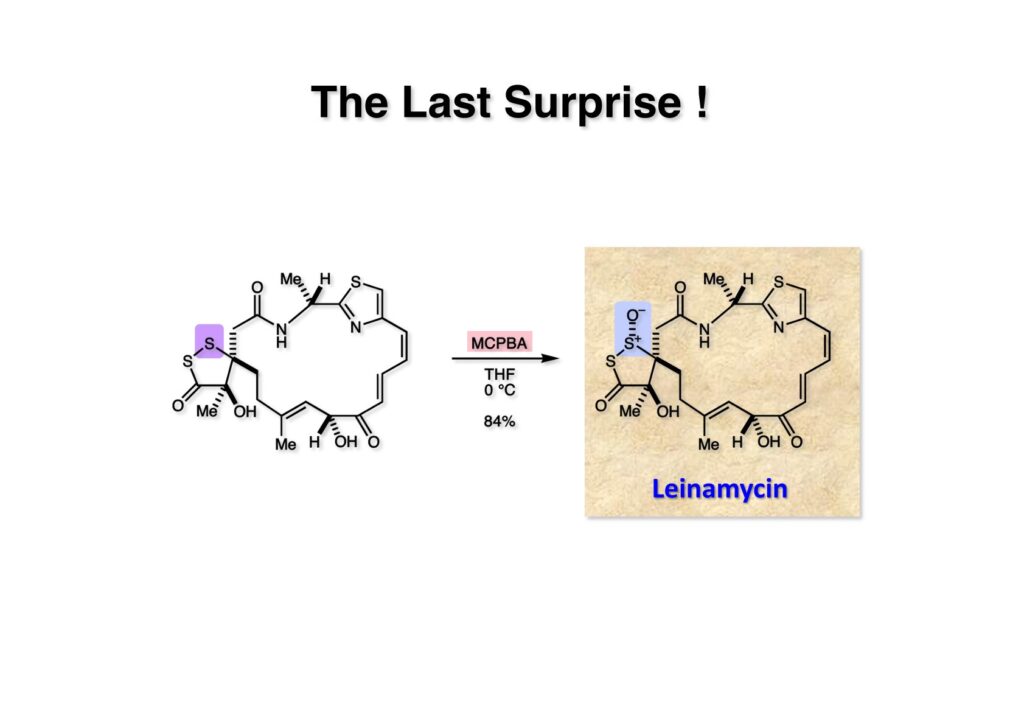 採集段階は硫黄の酸化なので有るが、立体化学をどう制御しようかということは予測ができなくて心配だった。うまくいかなければ不斉酸化剤を使うか、使うとしたらどんな試薬を使うべきか、などなど色々考えていた。ところが神田さんからMCPBA酸化したらleinamycinが得られたという連絡があり、「案ずるより産むが易し」とはまさにこの事か、と。振り返ってみると、色々なことを考えさせてくれた面白い全合成だったなーと感慨も一入である。
採集段階は硫黄の酸化なので有るが、立体化学をどう制御しようかということは予測ができなくて心配だった。うまくいかなければ不斉酸化剤を使うか、使うとしたらどんな試薬を使うべきか、などなど色々考えていた。ところが神田さんからMCPBA酸化したらleinamycinが得られたという連絡があり、「案ずるより産むが易し」とはまさにこの事か、と。振り返ってみると、色々なことを考えさせてくれた面白い全合成だったなーと感慨も一入である。