Mitomycin C(MMC)は1955年に北里研究所で発見され、1958年に協和発酵で結晶として単離された抗がん剤で、現在も臨床的に使われている。低分子で合成がそれほど難しくないと思いがちで数々の合成研究が報告されてきたが、現在までに恩師の岸義人先生とライス大学在籍時の私の研究室でしか全合成に成功していない。1977年1月20日にハーバード大学で博士口述試験を終えた翌日に岸先生のオフィスに呼ばれ、MMCの全合成を3名のポスドクがやっているが、なかなか終わらないので、とにかく終わっらせてほしいと頼まれた。半年ほど頑張って全合成は完成したが、多段階合成でとにかく終わらせた、という感じだった。Mitomycin A(MMA)はキノン部分のアミノ基がメトキシ基になっている化合物で、アンモニアを通ずると簡単にMMCに変換できる。最初にMMAを合成した時は1.5 mgの中間体から5段階で到達したのだが、MMAのキノン骨格を持つ化合物は綺麗な紫色なので、TLC上で追跡するのは簡単だった。合成されたMMAはおそらく0.5 mg以下だったと思うが、ハーバードの80 MHzのNMRではメチル基のシグナルが3本天然物と一致したのと、TLCも同じRfだったことで合成ができたことが分かった。勿論、最終的にはスケールアプして論文を報告している。MMCの色は濃紺でMMAの高貴な色の方が感じが良かった。ま、とにかくライス大学に移ってからも、もっと効率的な合成ルートがあるはずだ、と思いつつ、なかなか全合成に着手したいと思うようなアイデアが浮かばなかったが、「マイトマイシン転位」が発見されたおかげで効率的な全合成を完成させる事ができた。南開大学出身の大学院1年生だったLihu Yangが一人でやってのけた研究である。
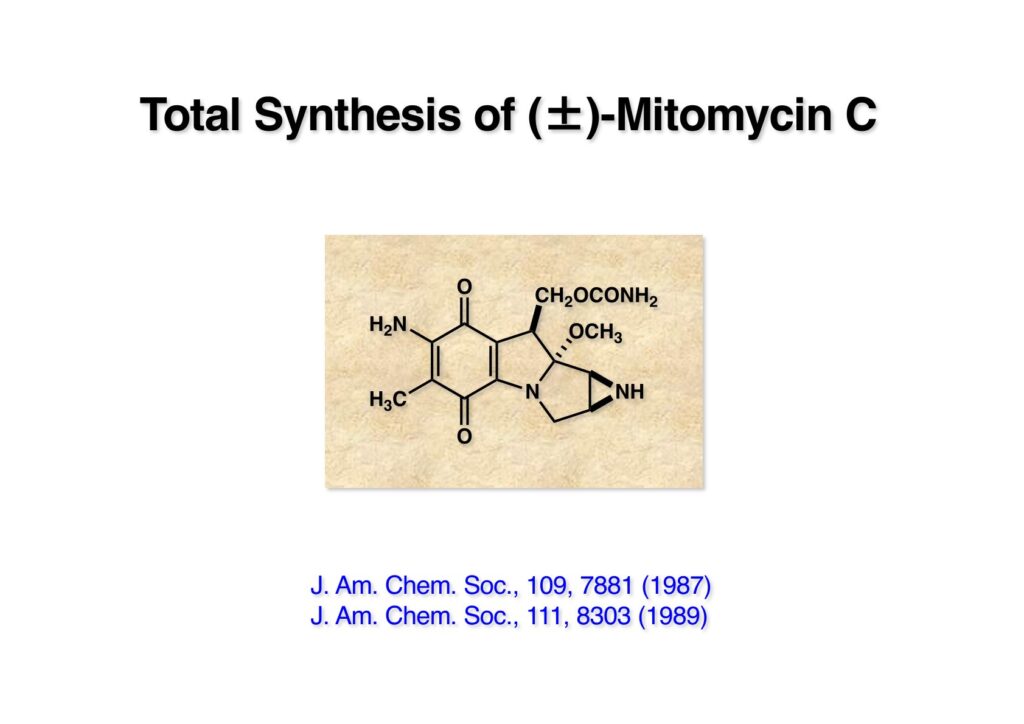
“Total Synthesis of (±)Mitomycins via Isomitomycin A,” T. Fukuyama and L.-H. Yang, J. Am. Chem. Soc., 109, 7881 (1987).
“Practical Total Synthesis of (±)-Mitomycin C,” T. Fukuyama and L.-H. Yang, J. Am. Chem. Soc., 111, 8303 (1989).
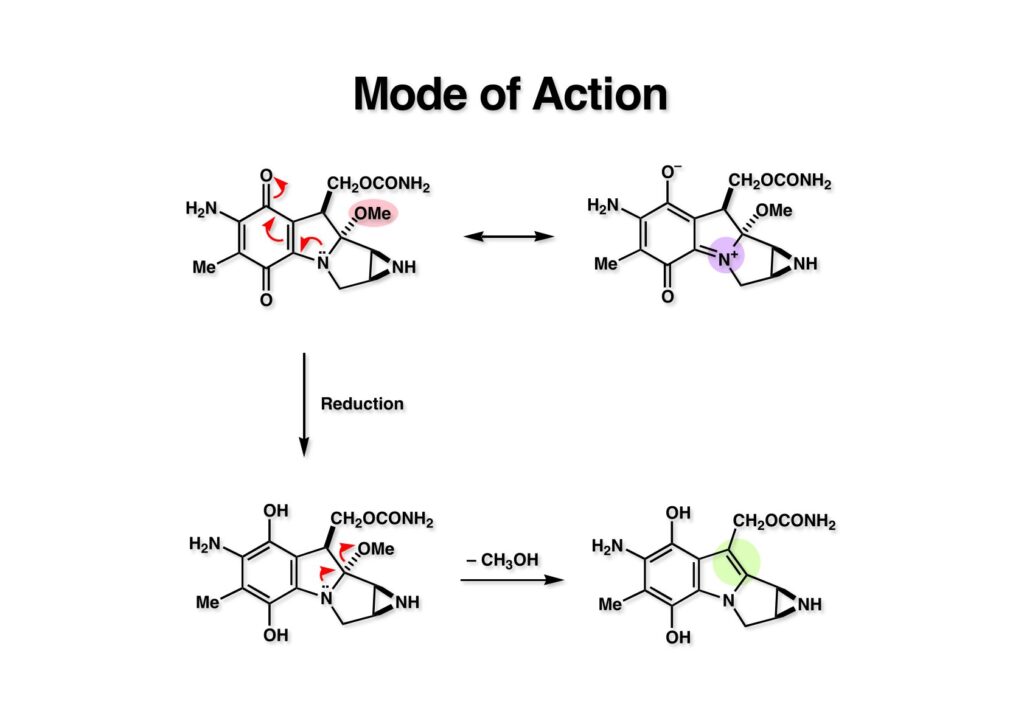 MMC (1-1) が天然に存在するほど安定なのはキノン構造を持っているからで、キノンに結合した三級アミンはvinylogous amideとなっているので孤立電子対によるMeO基の追い出しには効果的でない。ところが生体内でMMCが還元されて芳香環 (2-1) になると、たちまち脱メタノール化が進行してインドール (2-2) になってしまう。
MMC (1-1) が天然に存在するほど安定なのはキノン構造を持っているからで、キノンに結合した三級アミンはvinylogous amideとなっているので孤立電子対によるMeO基の追い出しには効果的でない。ところが生体内でMMCが還元されて芳香環 (2-1) になると、たちまち脱メタノール化が進行してインドール (2-2) になってしまう。
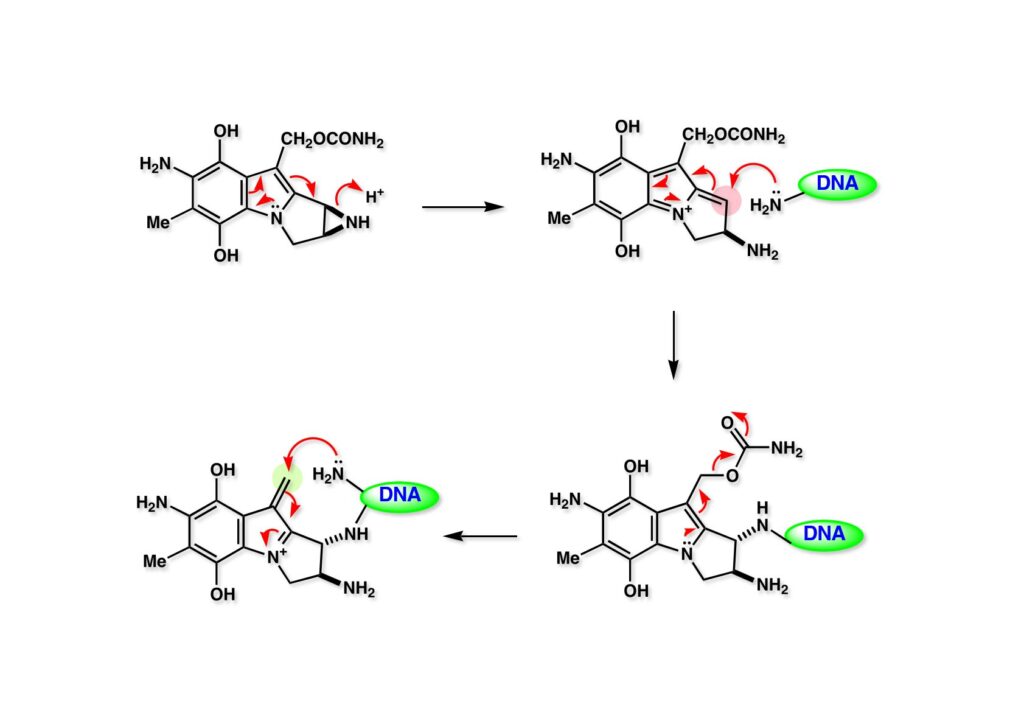 インドール (1-1) のアジリジン窒素原子がプロトン化されるとアジリジン環の開裂が即座に進行し、生じた活性種 (1-2) がDNAのC-G, G-Cペアのグアニンのアミノ基を攻撃する。次いで、インドール上の窒素の孤立電子対の押し出しによってカーバメートが脱離し、対面のグアニンを攻撃する。
インドール (1-1) のアジリジン窒素原子がプロトン化されるとアジリジン環の開裂が即座に進行し、生じた活性種 (1-2) がDNAのC-G, G-Cペアのグアニンのアミノ基を攻撃する。次いで、インドール上の窒素の孤立電子対の押し出しによってカーバメートが脱離し、対面のグアニンを攻撃する。
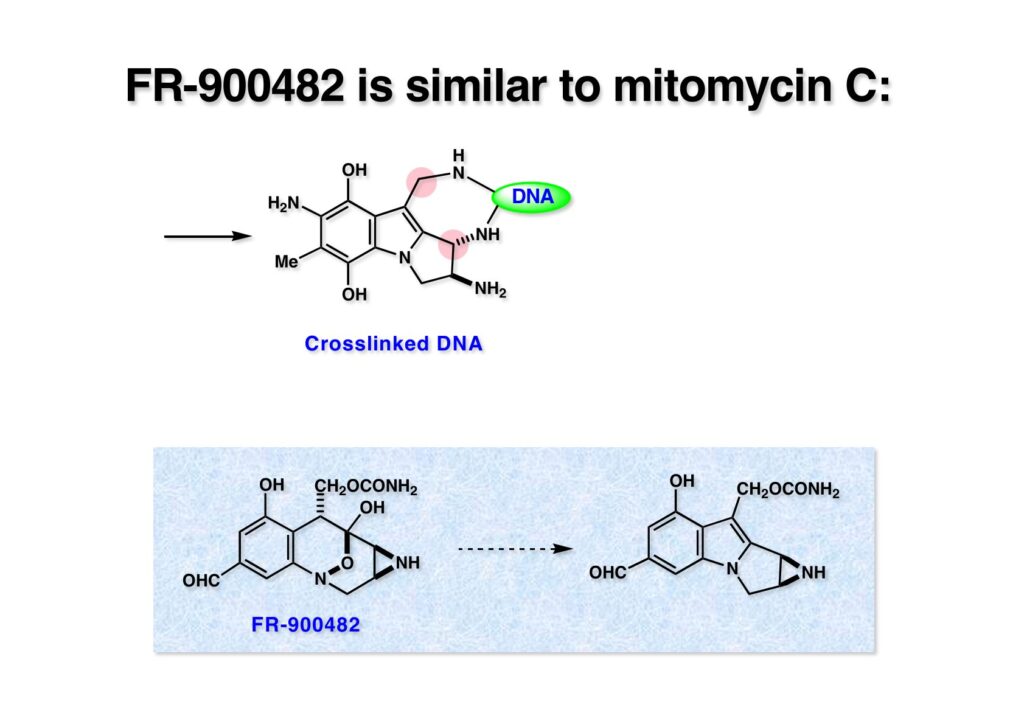 こうしてDNAの二重鎖が架橋されて複製不能になるのがMMCの作用機構である。このメカニズムの化学的証明はニューヨーク市大のMaria Tomaszとコロンビア大学の中西香爾先生の共同研究で行われた。Greg Verdineは中西研の院生としてこの研究に従事した。後に藤沢薬品で単離構造決定されたFR-900482もMMCと同様の作用機構でDNAをcross-linkingすることが知られている。この化合物の全合成も私たちの研究室で達成したので後述することになる。
こうしてDNAの二重鎖が架橋されて複製不能になるのがMMCの作用機構である。このメカニズムの化学的証明はニューヨーク市大のMaria Tomaszとコロンビア大学の中西香爾先生の共同研究で行われた。Greg Verdineは中西研の院生としてこの研究に従事した。後に藤沢薬品で単離構造決定されたFR-900482もMMCと同様の作用機構でDNAをcross-linkingすることが知られている。この化合物の全合成も私たちの研究室で達成したので後述することになる。
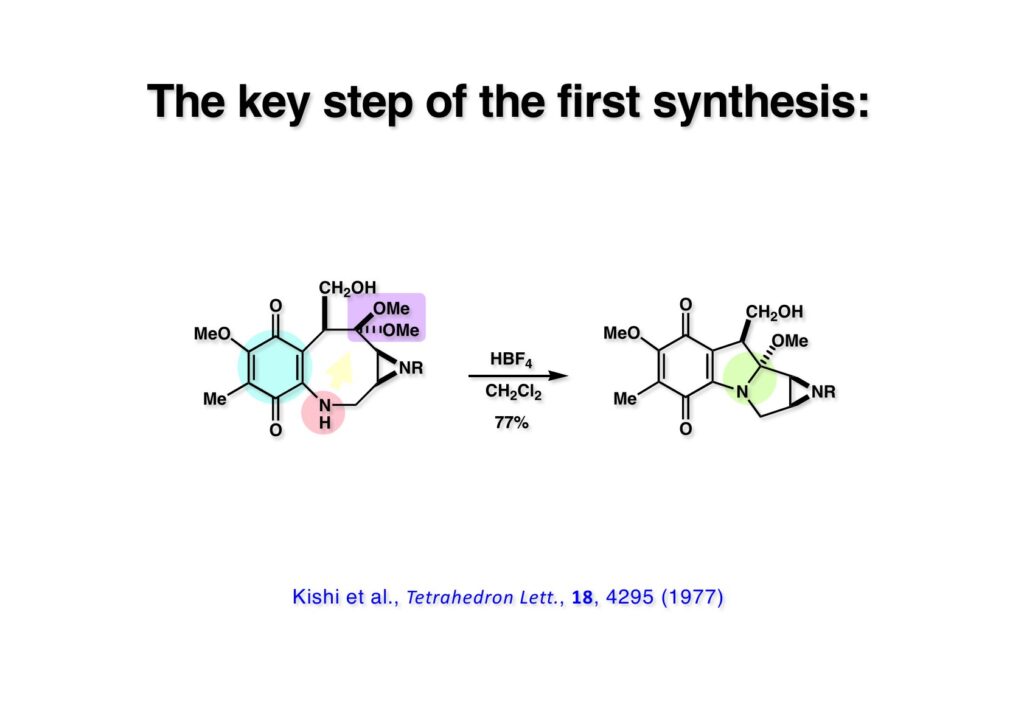 キノン骨格を持っていなければマイトマイシンとして安定に存在できないので、岸先生のアイデアは8員環キノン体 (1-1) で渡環反応を行うことでMMA骨格に導こうとした。この反応は生成物も酸に不安定なので綱渡り的な条件が必要となる。岸先生がUCLAのMike Jungから聞いたのだと思うが、D. H. R. Bartonが Ph3CBF4を使ってケタールのhydride abstractionによる脱ケタール反応があるから、ジメチルケタール (1-1) のメチル基のHを引き抜いて渡環反応をやろうというアイデアを出された。市販の試薬を使って1 mgほどのスケールで反応を行ったところ、比較的きれいにMMA骨格を持つ化合物が得られた。しかし、こんな微妙な反応がこのスケールでうまく進行するはずがないと思い、HBF4を少量使ってみたら同様の結果が得られた。
キノン骨格を持っていなければマイトマイシンとして安定に存在できないので、岸先生のアイデアは8員環キノン体 (1-1) で渡環反応を行うことでMMA骨格に導こうとした。この反応は生成物も酸に不安定なので綱渡り的な条件が必要となる。岸先生がUCLAのMike Jungから聞いたのだと思うが、D. H. R. Bartonが Ph3CBF4を使ってケタールのhydride abstractionによる脱ケタール反応があるから、ジメチルケタール (1-1) のメチル基のHを引き抜いて渡環反応をやろうというアイデアを出された。市販の試薬を使って1 mgほどのスケールで反応を行ったところ、比較的きれいにMMA骨格を持つ化合物が得られた。しかし、こんな微妙な反応がこのスケールでうまく進行するはずがないと思い、HBF4を少量使ってみたら同様の結果が得られた。
 私はMMCのことを「飛んで火に入る夏の虫的化合物」と読んでいる。「あーあの有名なマイトマイシンCか、どれどれ、小さくて簡単そうな化合物だねー。これならオイラでも全合成できそうだ。」なんちゃって、結構たくさんのケミストが全合成に挑戦したが、今までに岸先生と吾輩だけが成功している。例えば、かの有名なDanishefsky先生は20年間院生とポスドクを使ってMMCの全合成に挑戦したが、ついに完成しなかった。最後の方の論文に、「MMCの合成は卵の殻の上を歩くようなものだ」と、その難しさに嘆いておられた。岸先生もポスドク4名(4人目が私)を駆使して3年間で何とか合成に成功されたが、2,6-dimethoxytolueneから45段階、通算収率0.2%という多段階合成であった。ここでは当研究室の二つの全合成を記述するが、中国の南開大学出身の大学院1年生であったLihu Yang(楊立虎)君が1年半で仕上げたものだ。二つの論文の発表時期に2年の間隔があるのは、最初の合成を1年で終え、その改良合成を半年で終えたので、改良合成はJACSにフルペーパーで出そうと思って机上に置いておいたのであるが、段々フルペーパーを書くのが面倒になり、結局最初のとはかなりルートが違うので2年後にJACSのコミュニケーションで出したというのが真相である。Yang君は私の学生の中でも1、2を争う極めて優秀な学生だったが、すぐに手抜きをしたがるので、隣でまだ時々実験をやっていた私がしょっちゅう注意していた。まだグリーンカードを持っていないと製薬会社は面接もしてくれない頃に、Merck社のexecutive directorをやっていたJim Heck (Woodward研出身)にLihuを面接してくれないかと頼んだ。その時に私が書いた推薦状に、「私が岸研に居た時に岸先生がどれくらい助かったかは、Lihuが私の研究室に居ることでよく分かる」「Lihuは私と同じくらい優秀だ(But, to tell you the truth, I am a little bit better than him.)」というような文面であったが、Jimは面白がって同僚にコピーを回覧したそうだ。勿論、LihuはMerck社に就職でき、後にdirectorに出世した。早期退職後はベルギーを拠点にウロウロしているようだ(仕事内容は知らない)。
私はMMCのことを「飛んで火に入る夏の虫的化合物」と読んでいる。「あーあの有名なマイトマイシンCか、どれどれ、小さくて簡単そうな化合物だねー。これならオイラでも全合成できそうだ。」なんちゃって、結構たくさんのケミストが全合成に挑戦したが、今までに岸先生と吾輩だけが成功している。例えば、かの有名なDanishefsky先生は20年間院生とポスドクを使ってMMCの全合成に挑戦したが、ついに完成しなかった。最後の方の論文に、「MMCの合成は卵の殻の上を歩くようなものだ」と、その難しさに嘆いておられた。岸先生もポスドク4名(4人目が私)を駆使して3年間で何とか合成に成功されたが、2,6-dimethoxytolueneから45段階、通算収率0.2%という多段階合成であった。ここでは当研究室の二つの全合成を記述するが、中国の南開大学出身の大学院1年生であったLihu Yang(楊立虎)君が1年半で仕上げたものだ。二つの論文の発表時期に2年の間隔があるのは、最初の合成を1年で終え、その改良合成を半年で終えたので、改良合成はJACSにフルペーパーで出そうと思って机上に置いておいたのであるが、段々フルペーパーを書くのが面倒になり、結局最初のとはかなりルートが違うので2年後にJACSのコミュニケーションで出したというのが真相である。Yang君は私の学生の中でも1、2を争う極めて優秀な学生だったが、すぐに手抜きをしたがるので、隣でまだ時々実験をやっていた私がしょっちゅう注意していた。まだグリーンカードを持っていないと製薬会社は面接もしてくれない頃に、Merck社のexecutive directorをやっていたJim Heck (Woodward研出身)にLihuを面接してくれないかと頼んだ。その時に私が書いた推薦状に、「私が岸研に居た時に岸先生がどれくらい助かったかは、Lihuが私の研究室に居ることでよく分かる」「Lihuは私と同じくらい優秀だ(But, to tell you the truth, I am a little bit better than him.)」というような文面であったが、Jimは面白がって同僚にコピーを回覧したそうだ。勿論、LihuはMerck社に就職でき、後にdirectorに出世した。早期退職後はベルギーを拠点にウロウロしているようだ(仕事内容は知らない)。
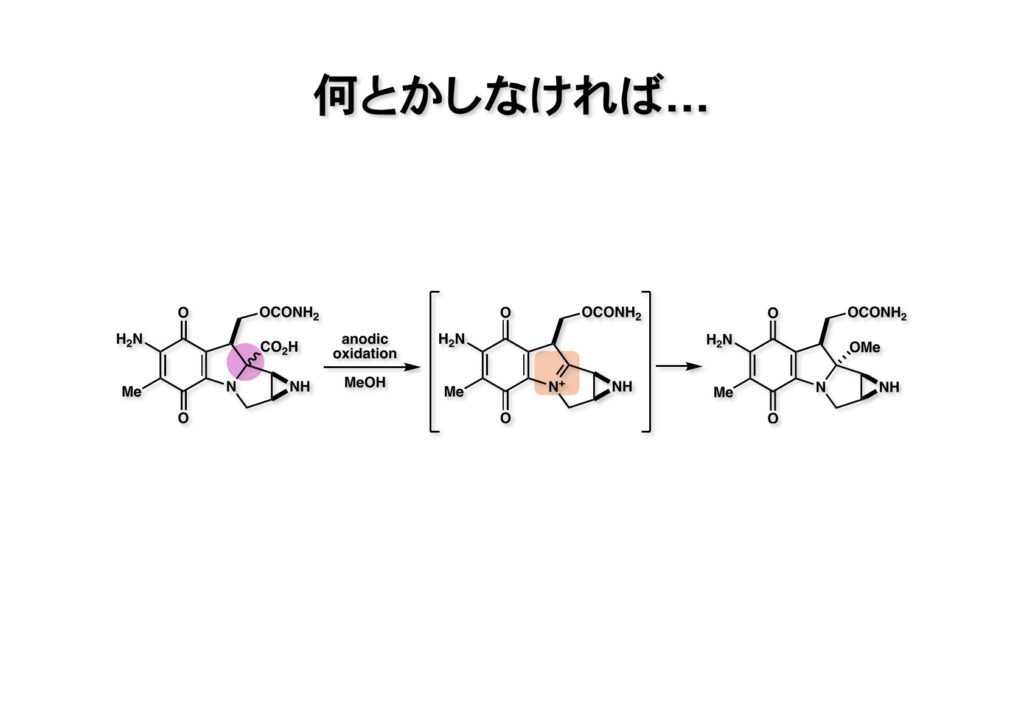 このアイデアはただの机上のプランで、MMCを効率的に合成するためには (1-1) のようなカルボン酸をメタノール中で陽極酸化 (anodic oxidation) するくらいしか方法はないのではないか、と思っていた。(1-2) が生成すれば、脱プロトン化する前にメタノールが立体障害の少ないα面から攻撃してMMC (1-3) が生成するはずである。このアイデアは実行に移さなかったが、成功確率は高いと思う。
このアイデアはただの机上のプランで、MMCを効率的に合成するためには (1-1) のようなカルボン酸をメタノール中で陽極酸化 (anodic oxidation) するくらいしか方法はないのではないか、と思っていた。(1-2) が生成すれば、脱プロトン化する前にメタノールが立体障害の少ないα面から攻撃してMMC (1-3) が生成するはずである。このアイデアは実行に移さなかったが、成功確率は高いと思う。
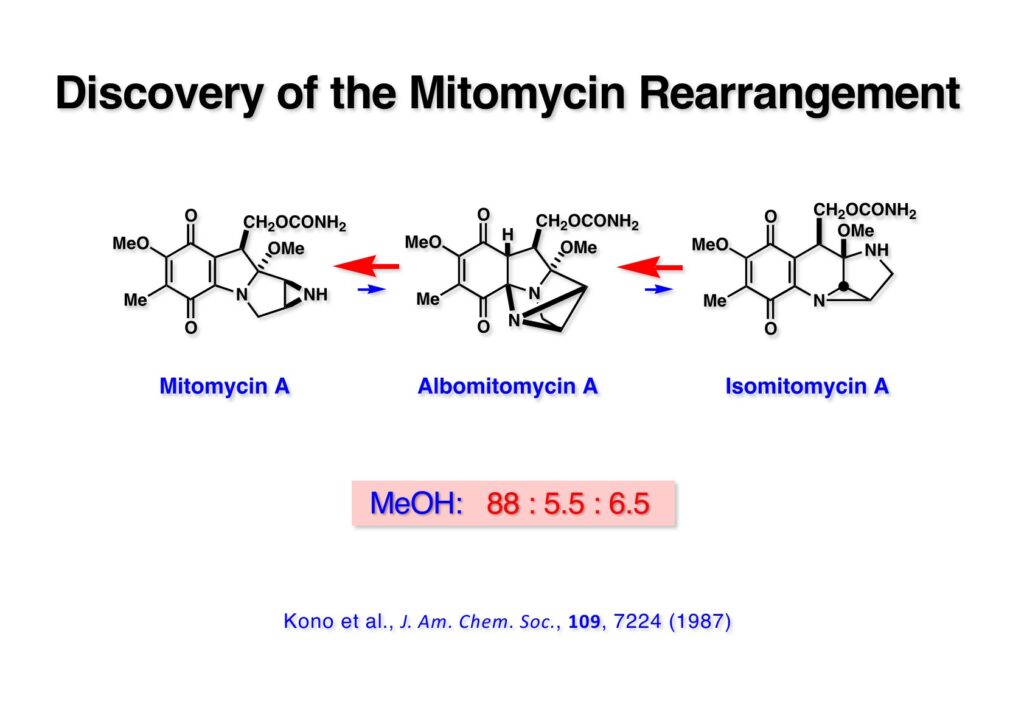 1985年の年末に三共株式会社の杉村征夫さんからクリスマスカードが送られてきて、カードの間にその年の天然有機化合物討論会の講演要旨が挟まっていた。杉村さんは毎年天然物討論会に出席した同僚の要らなくなった要旨集を私に郵送してくれていた。その年は要旨集が手に入らなかったけれど、面白い講演があったのでそのページをコピーして送って下さったのだ。それを見た瞬間「ガーン」と頭を打たれたような衝撃が走った。何と!協和発酵の研究者が「マイトマイシン転位」を報告していたのだ!彼らはisomitomycin A (IMMA, 1-3) がalbomitomycin A (AMMA, 1-2) を経由してmitomycin A (MMA, 1-1) に変換でき、平衡混合物の比は左から 88:5.5:6.5 となっていた。
1985年の年末に三共株式会社の杉村征夫さんからクリスマスカードが送られてきて、カードの間にその年の天然有機化合物討論会の講演要旨が挟まっていた。杉村さんは毎年天然物討論会に出席した同僚の要らなくなった要旨集を私に郵送してくれていた。その年は要旨集が手に入らなかったけれど、面白い講演があったのでそのページをコピーして送って下さったのだ。それを見た瞬間「ガーン」と頭を打たれたような衝撃が走った。何と!協和発酵の研究者が「マイトマイシン転位」を報告していたのだ!彼らはisomitomycin A (IMMA, 1-3) がalbomitomycin A (AMMA, 1-2) を経由してmitomycin A (MMA, 1-1) に変換でき、平衡混合物の比は左から 88:5.5:6.5 となっていた。
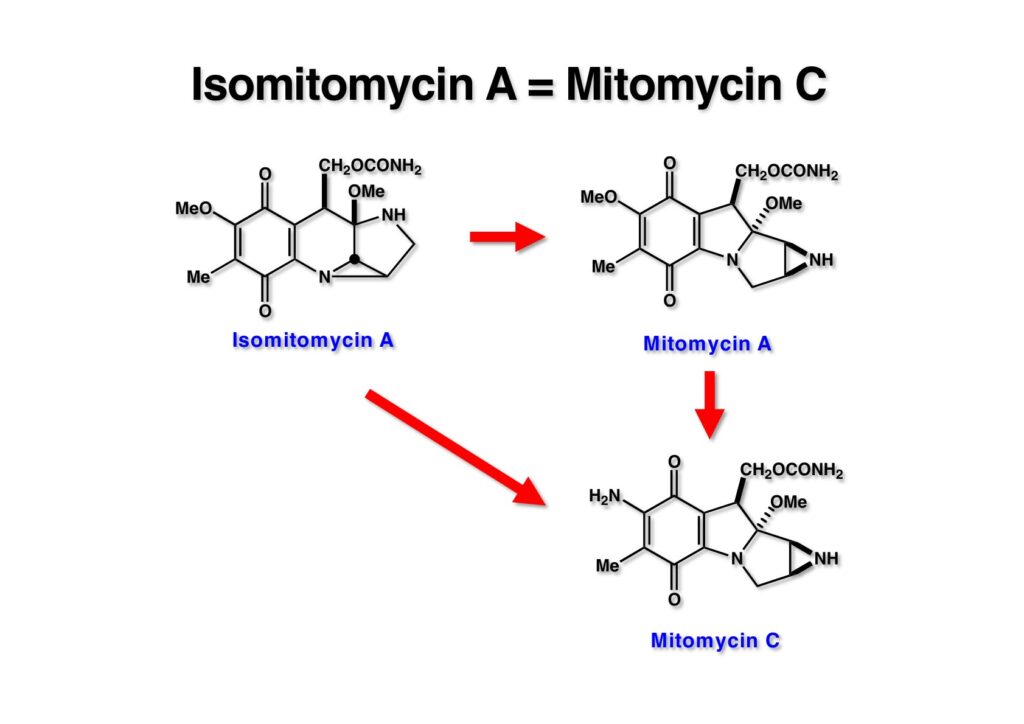 すなわち、IMMA (1-1) を合成すればMMA (1-2) ひいてはMMC (2-1) に変換できるということである。Isomitomycin Aではキノンを芳香環にしておいても脱メタノールが起きないし、MeO基は橋頭位に結合しているので容易には脱離しない筈である。ところが、後になって分かったことだが、mitomycin Aよりもisomitomycin Aの方が酸性条件で安定だと推測していたのに、後者の方が不安定であることが意外であった。
すなわち、IMMA (1-1) を合成すればMMA (1-2) ひいてはMMC (2-1) に変換できるということである。Isomitomycin Aではキノンを芳香環にしておいても脱メタノールが起きないし、MeO基は橋頭位に結合しているので容易には脱離しない筈である。ところが、後になって分かったことだが、mitomycin Aよりもisomitomycin Aの方が酸性条件で安定だと推測していたのに、後者の方が不安定であることが意外であった。
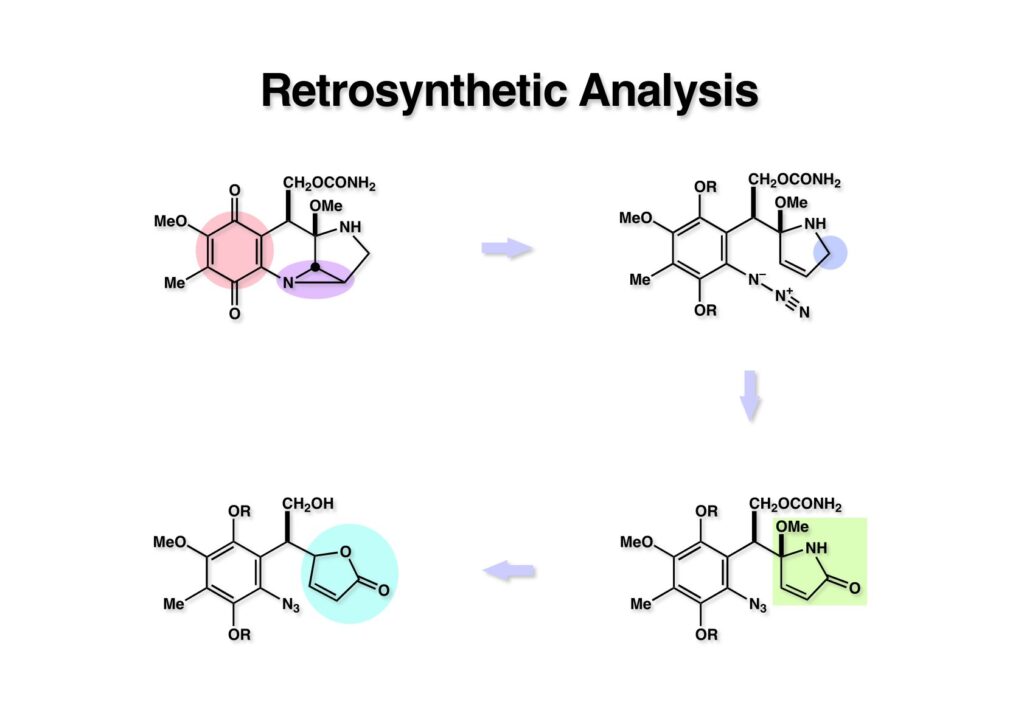 早速IMMA (1-1) の逆合成解析に着手した。まず、キノンは芳香環として保護をし、アジリジンの構築はオレフィンとアジドの1,3-双極子付加とそれに続く窒素の脱離によって構築することとして (1-2) に導いた。この反応は時としてイミンが主生成物になる可能性もあるが、その時はその時でまた考えることにした。(1-2) の構造ではすぐにメタノールが脱離してピロールになってしまうので、それを防ぐためにカルボニル基を環上に配置して (2-2) とした。(2-2) のピロリノン環では炭素-炭素結合が作りにくいので、より簡単なブテノライド (2-1) をピロリノンの前駆体とした。
早速IMMA (1-1) の逆合成解析に着手した。まず、キノンは芳香環として保護をし、アジリジンの構築はオレフィンとアジドの1,3-双極子付加とそれに続く窒素の脱離によって構築することとして (1-2) に導いた。この反応は時としてイミンが主生成物になる可能性もあるが、その時はその時でまた考えることにした。(1-2) の構造ではすぐにメタノールが脱離してピロールになってしまうので、それを防ぐためにカルボニル基を環上に配置して (2-2) とした。(2-2) のピロリノン環では炭素-炭素結合が作りにくいので、より簡単なブテノライド (2-1) をピロリノンの前駆体とした。
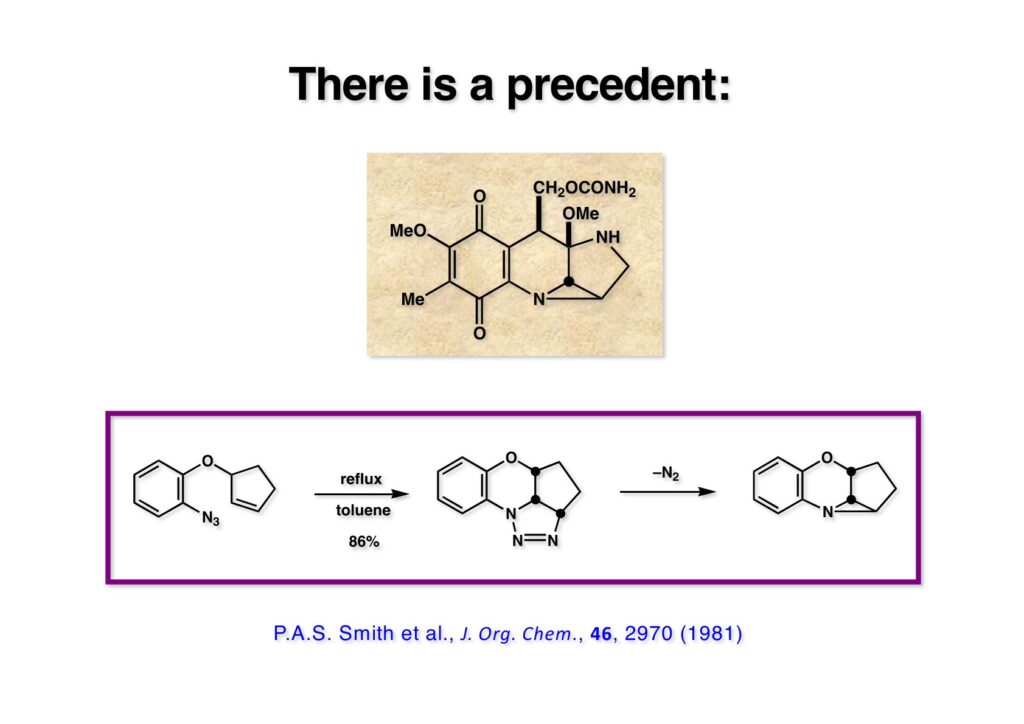 アジドとオレフィンの環化付加反応の例を探しに図書館に行き、Chemical Abstractsを調べたところ、 (2-1) をトルエン中で還流すると高収率でアジリジン (2-3) が生成するという報告があった。今ならSciFinderであっという間に探せるのだが、昔はこれを探すのも容易ではなかった。とにかくこの論文を見た時にMMCの全合成は終わったような気になったのは確かである。
アジドとオレフィンの環化付加反応の例を探しに図書館に行き、Chemical Abstractsを調べたところ、 (2-1) をトルエン中で還流すると高収率でアジリジン (2-3) が生成するという報告があった。今ならSciFinderであっという間に探せるのだが、昔はこれを探すのも容易ではなかった。とにかくこの論文を見た時にMMCの全合成は終わったような気になったのは確かである。
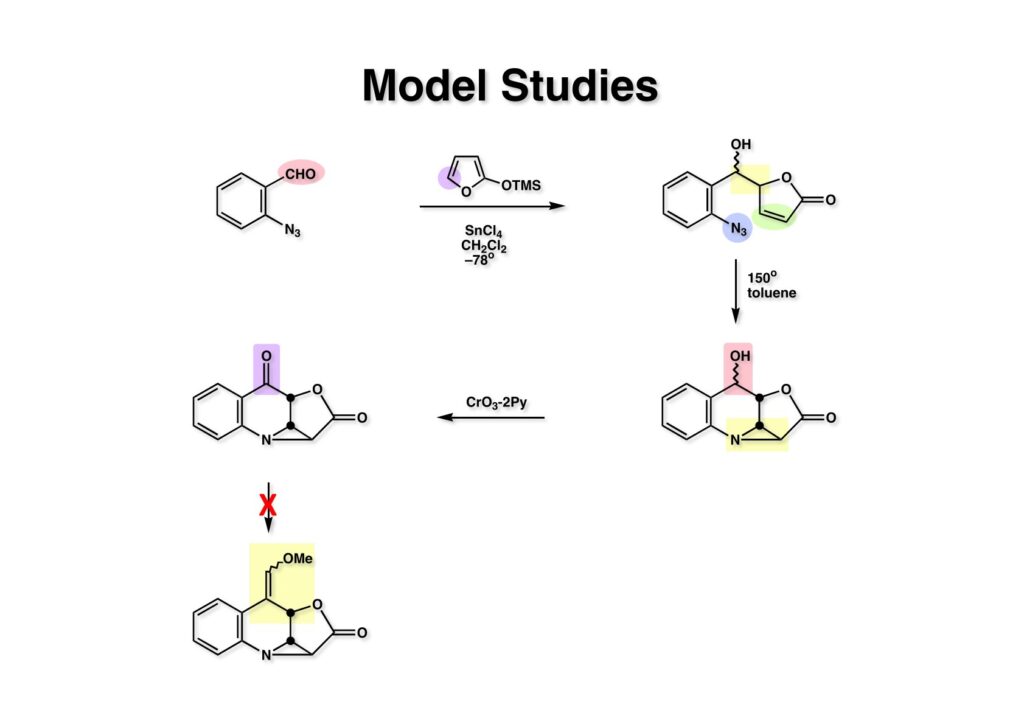 ジュネーブ大学のCharles Jefford教授は2-trimethylsilyloxyfuran (1-2) を用いて、種々のブテノライドの合成を報告していたので、その例にならって (1-1) への付加反応を行ったところ (1-3) が得られた。これを封管中で加熱したところ望むアジリジン (2-2) が得られた。次に、このアルコールを酸化してケトン (2-1) に変換した。次にWittig反応でケトンから1炭素増炭を試みたが、ラクトンの方がケトンよりも反応性が高くて望む化合物 (3-1) は得られなかった。そこで次の策として、少しアトムエコノミーの観点からはダサいけれど、カルコンへのMichael付加をやってみることにした。
ジュネーブ大学のCharles Jefford教授は2-trimethylsilyloxyfuran (1-2) を用いて、種々のブテノライドの合成を報告していたので、その例にならって (1-1) への付加反応を行ったところ (1-3) が得られた。これを封管中で加熱したところ望むアジリジン (2-2) が得られた。次に、このアルコールを酸化してケトン (2-1) に変換した。次にWittig反応でケトンから1炭素増炭を試みたが、ラクトンの方がケトンよりも反応性が高くて望む化合物 (3-1) は得られなかった。そこで次の策として、少しアトムエコノミーの観点からはダサいけれど、カルコンへのMichael付加をやってみることにした。
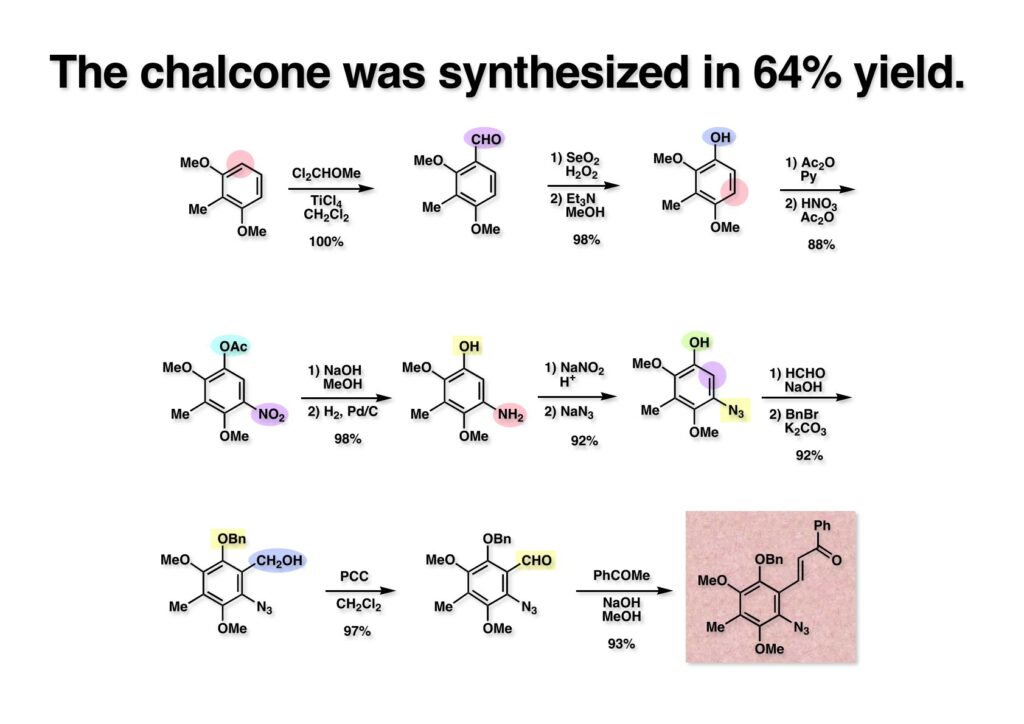 ここに示したカルコン (3-3) の合成は38歳の私が最後に行った実験である。市販の2,6-dimethoxytoluene (1-1) 25 gをFriedel-Crafts反応でアルデヒド (1-2) に変換した。Bicyclomycinの全合成の時に述べたが、このスケールでMCPBAを用いると後処理が面倒なので、アルデヒドと触媒量のSeO2のt-BuOH溶液に50度を超えないように30% H2O2を滴下していくとギ酸エステルが高収率で得られる。これを分液後に蒸留乾固し、精製することなくメタノール中でEt3Nを加えてフェノール (1-3) を得た。次に、フェノールをアセチル化して保護し、無水酢酸と硝酸の混合液でニトロ化すると (2-1) が得られる。アセテートはちょっとEt3N-MeOHの条件では落ちないので、普通に加水分解し、耐圧容器中でPd/Cを触媒に高圧水添すると短時間でアミン (2-2) 得られる。このアニリン (2-2) からアジド (2-3) への変換は非常に簡単で、まず常法でジアゾニウム塩を作り、それにNaN3を加えていくと窒素ガスがブクブクと出てくるが、泡が出終わったら反応終了である。フェノールのホルマリンによるヒドロキシメチル化は、位置選択的に反応させるのが特殊例を除いて困難であるが、この場合はオルト位が一箇所空いているだけなので簡単に生成物が得られる。フェノールのベンジル化は当然aliphatic alcoholが存在しても選択的に進行し (3-1) を得た。この頃になってくると不純物も目立つようになったが、スケールが多少大きいのでカラム精製はなるべく先送りするつもりだった。次ステップのアルコールのPCC酸化でアルデヒド (3-2) は得られたもののカラム精製したくなるくらい不純物が増加してきた。しかしカルコンまで到達してから精製する事にして、アルデヒド (3-2) とPhCOMeをメタノールに溶かして、攪拌しながらNaOH水溶液を加えていったところ、キラキラ光る結晶が現れてきた。反応終了後に結晶を濾過乾燥して収率を計算したところ、1週間かけて12段階で通算収率が49%であった。学生たちに私が昔は如何に実験の腕が良かったかを自慢するネタである。このカルコンを使って、合成を始めて1年と数日でMMCの全合成は完成した。Lihuがこのカルコンはどうやって合成したのか?って聞きにきたが、何しろ頭の中の常識を使って合成経路を考えたのと、少々忙しかったのと面倒臭がりのせいで、全く私の実験ノートに記録してなかった。慌ててルートを紙に書いてやり、適当に反応条件を教えてやったら3週間ほどで合成ルートが再現できた。Lihuは私に勝とうとして、同じスケールでカルコンを合成したが、通算収率は46%で軍配は私に上がった。
ここに示したカルコン (3-3) の合成は38歳の私が最後に行った実験である。市販の2,6-dimethoxytoluene (1-1) 25 gをFriedel-Crafts反応でアルデヒド (1-2) に変換した。Bicyclomycinの全合成の時に述べたが、このスケールでMCPBAを用いると後処理が面倒なので、アルデヒドと触媒量のSeO2のt-BuOH溶液に50度を超えないように30% H2O2を滴下していくとギ酸エステルが高収率で得られる。これを分液後に蒸留乾固し、精製することなくメタノール中でEt3Nを加えてフェノール (1-3) を得た。次に、フェノールをアセチル化して保護し、無水酢酸と硝酸の混合液でニトロ化すると (2-1) が得られる。アセテートはちょっとEt3N-MeOHの条件では落ちないので、普通に加水分解し、耐圧容器中でPd/Cを触媒に高圧水添すると短時間でアミン (2-2) 得られる。このアニリン (2-2) からアジド (2-3) への変換は非常に簡単で、まず常法でジアゾニウム塩を作り、それにNaN3を加えていくと窒素ガスがブクブクと出てくるが、泡が出終わったら反応終了である。フェノールのホルマリンによるヒドロキシメチル化は、位置選択的に反応させるのが特殊例を除いて困難であるが、この場合はオルト位が一箇所空いているだけなので簡単に生成物が得られる。フェノールのベンジル化は当然aliphatic alcoholが存在しても選択的に進行し (3-1) を得た。この頃になってくると不純物も目立つようになったが、スケールが多少大きいのでカラム精製はなるべく先送りするつもりだった。次ステップのアルコールのPCC酸化でアルデヒド (3-2) は得られたもののカラム精製したくなるくらい不純物が増加してきた。しかしカルコンまで到達してから精製する事にして、アルデヒド (3-2) とPhCOMeをメタノールに溶かして、攪拌しながらNaOH水溶液を加えていったところ、キラキラ光る結晶が現れてきた。反応終了後に結晶を濾過乾燥して収率を計算したところ、1週間かけて12段階で通算収率が49%であった。学生たちに私が昔は如何に実験の腕が良かったかを自慢するネタである。このカルコンを使って、合成を始めて1年と数日でMMCの全合成は完成した。Lihuがこのカルコンはどうやって合成したのか?って聞きにきたが、何しろ頭の中の常識を使って合成経路を考えたのと、少々忙しかったのと面倒臭がりのせいで、全く私の実験ノートに記録してなかった。慌ててルートを紙に書いてやり、適当に反応条件を教えてやったら3週間ほどで合成ルートが再現できた。Lihuは私に勝とうとして、同じスケールでカルコンを合成したが、通算収率は46%で軍配は私に上がった。
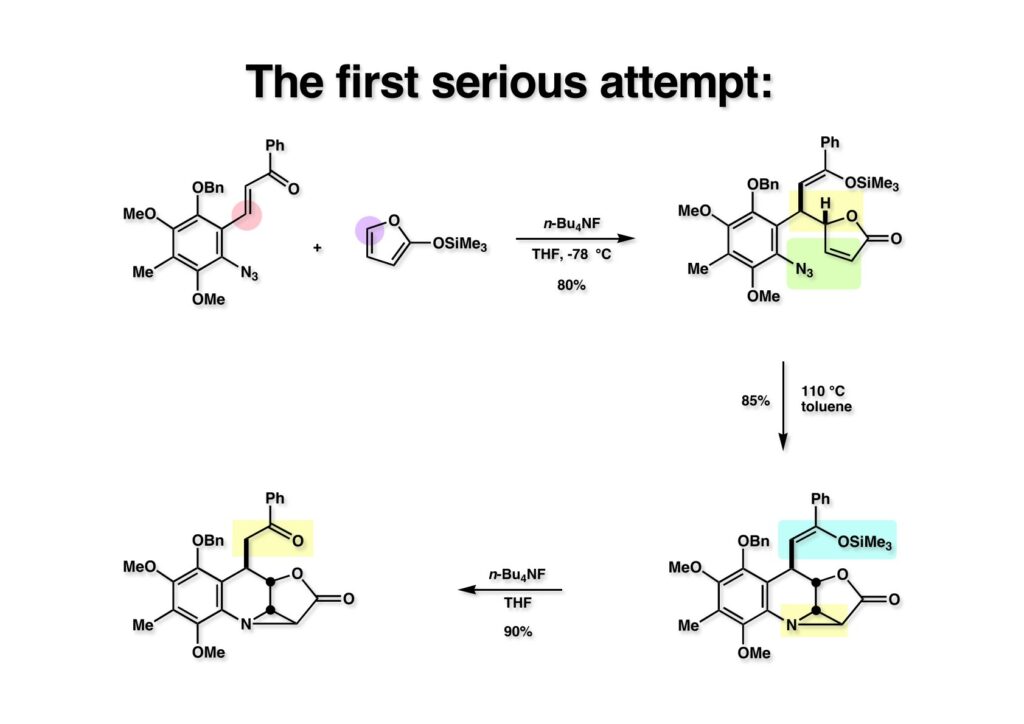 カルコン (1-1) と2-TMSO-furan (1-2) のTHF溶液に低温下TBAFを加えると高収率で付加体 (1-3) が得られた。NMRでは3:1の混合物だったのでブテノライドがエピメリ化したのだと思った。これをトルエン中で環流すると単一物 (2-2) が生成した。ここでもブテノライドが異性化して遷移状態のエネルギーが低い方が環化したものと解釈していた。この時点で酸に弱いシリルエノールエーテルを除去してケトン (2-1) に変換した。
カルコン (1-1) と2-TMSO-furan (1-2) のTHF溶液に低温下TBAFを加えると高収率で付加体 (1-3) が得られた。NMRでは3:1の混合物だったのでブテノライドがエピメリ化したのだと思った。これをトルエン中で環流すると単一物 (2-2) が生成した。ここでもブテノライドが異性化して遷移状態のエネルギーが低い方が環化したものと解釈していた。この時点で酸に弱いシリルエノールエーテルを除去してケトン (2-1) に変換した。
 アジリジン (1-1) をEtOHで開裂して (1-2) にしたが、水で開館してメシレートにすると、すぐに閉館してアジリジンに戻ることは確認済みで、EtO基は後で除去可能なアルコールにする予定だった。(1-2) のラクトンはアンモニアで開裂してアミド (2-2) を与えた。ここで水酸基を酸化してケトン (2-1) に変換しようとしたが、何をやっても分解してしまい望むケトンは得られなかった。当然、(1-1) の段階でラクトンの加アンモニア分解は行っているはずで、多分解列しなかったのだと思う。とにかく、(1-2) のアミンが全く保護できなかったのが痛かった。今から思えば、(2-2) でエチル基の代わりに水酸基にして何とかアジリジンに戻してからケトンへの酸化を試みれば良かったかもしれない。ま、それはそれで回りくどいルートになっていただろうけど。結局、フランの5位にヘテロ原子を導入することで、後の酸化を不必要にするアイデアを実行することにした。
アジリジン (1-1) をEtOHで開裂して (1-2) にしたが、水で開館してメシレートにすると、すぐに閉館してアジリジンに戻ることは確認済みで、EtO基は後で除去可能なアルコールにする予定だった。(1-2) のラクトンはアンモニアで開裂してアミド (2-2) を与えた。ここで水酸基を酸化してケトン (2-1) に変換しようとしたが、何をやっても分解してしまい望むケトンは得られなかった。当然、(1-1) の段階でラクトンの加アンモニア分解は行っているはずで、多分解列しなかったのだと思う。とにかく、(1-2) のアミンが全く保護できなかったのが痛かった。今から思えば、(2-2) でエチル基の代わりに水酸基にして何とかアジリジンに戻してからケトンへの酸化を試みれば良かったかもしれない。ま、それはそれで回りくどいルートになっていただろうけど。結局、フランの5位にヘテロ原子を導入することで、後の酸化を不必要にするアイデアを実行することにした。
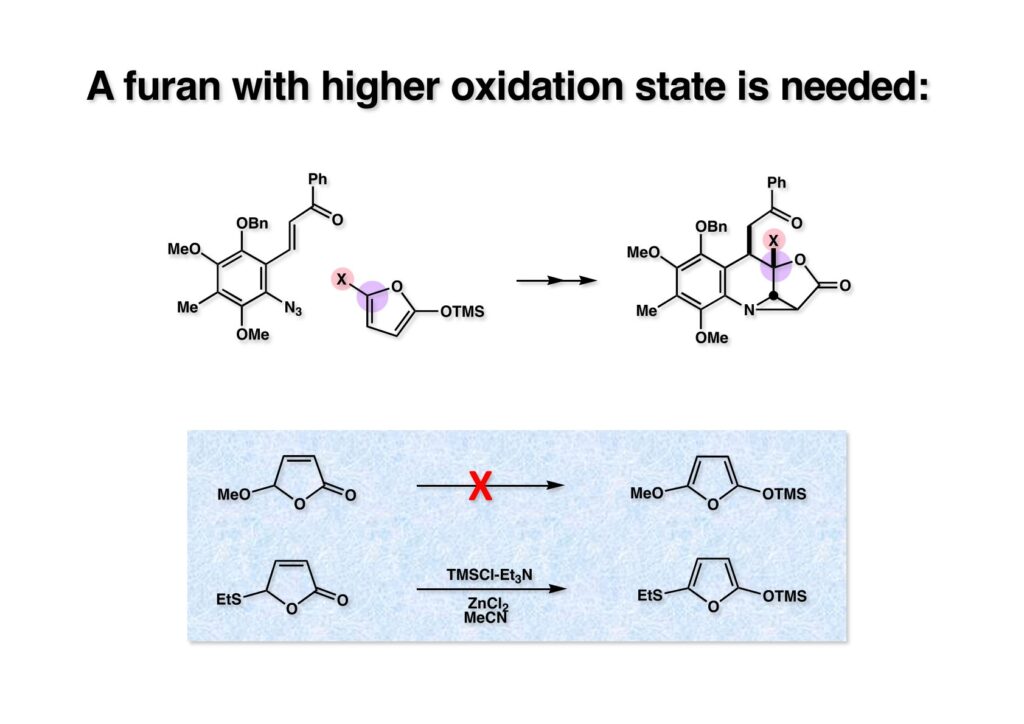 (1-2) のようなヘテロ原子Xを5位に入れておけば、(1-3) になる筈であり、前述のような困難な酸化をする必要がなくなる。ブテノライドから2-TMSO-furanを合成するには、アセトニトリル中で、TMSCl、Et3N、ZnCl2と反応させれば良い。ところが、5-methoxybutenolide (2-1) を同条件に付すと、まるで望むフラン体 (2-2) は得られなかった。そこで5-ethylthiobutenolide (3-1) を5-hydroxybutenolide (フルフラールをエタノール中で光酸化して合成)から合成し、同条件に付すと、容易に (3-2) が得られた。(2-2) が得られないのが腑に落ちなかったので、(2-1) と (3-1) の混合物を同条件に付したところ、(3-2) は得られたが、(2-2) は全く得られなかった。一体全体どうなってるの??
(1-2) のようなヘテロ原子Xを5位に入れておけば、(1-3) になる筈であり、前述のような困難な酸化をする必要がなくなる。ブテノライドから2-TMSO-furanを合成するには、アセトニトリル中で、TMSCl、Et3N、ZnCl2と反応させれば良い。ところが、5-methoxybutenolide (2-1) を同条件に付すと、まるで望むフラン体 (2-2) は得られなかった。そこで5-ethylthiobutenolide (3-1) を5-hydroxybutenolide (フルフラールをエタノール中で光酸化して合成)から合成し、同条件に付すと、容易に (3-2) が得られた。(2-2) が得られないのが腑に落ちなかったので、(2-1) と (3-1) の混合物を同条件に付したところ、(3-2) は得られたが、(2-2) は全く得られなかった。一体全体どうなってるの??
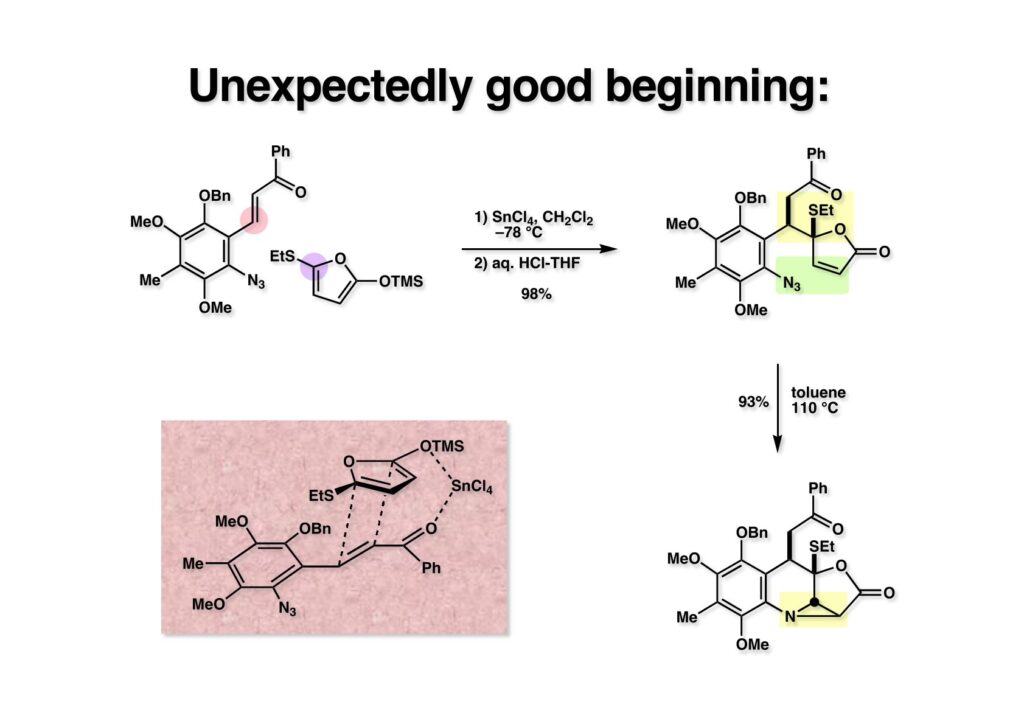 (1-2) のカルコン (1-1) への付加はTBAFでは全く進行しなかったので、ルイス酸を使う向山反応の条件を用いたところ反応は進行し、粗生成物を希塩酸で加水分解することで付加体 (1-3) を得た。TLC上ではワンスポットであったがNMRを見てみると3:1の混合物であった。これをトルエン中還流すると単一物のアジリジン (2-2) が得られてきた。ここではブテノライドが異性化する筈がないので、(1-2) はアトロプ異性体であることが分かった。(2-2) の立体化学はNOE実験で決定し、これにより、Diels-Alder反応のendo-型付加反応様の遷移状態 (2-1) を取ったのではないかと推測している。
(1-2) のカルコン (1-1) への付加はTBAFでは全く進行しなかったので、ルイス酸を使う向山反応の条件を用いたところ反応は進行し、粗生成物を希塩酸で加水分解することで付加体 (1-3) を得た。TLC上ではワンスポットであったがNMRを見てみると3:1の混合物であった。これをトルエン中還流すると単一物のアジリジン (2-2) が得られてきた。ここではブテノライドが異性化する筈がないので、(1-2) はアトロプ異性体であることが分かった。(2-2) の立体化学はNOE実験で決定し、これにより、Diels-Alder反応のendo-型付加反応様の遷移状態 (2-1) を取ったのではないかと推測している。
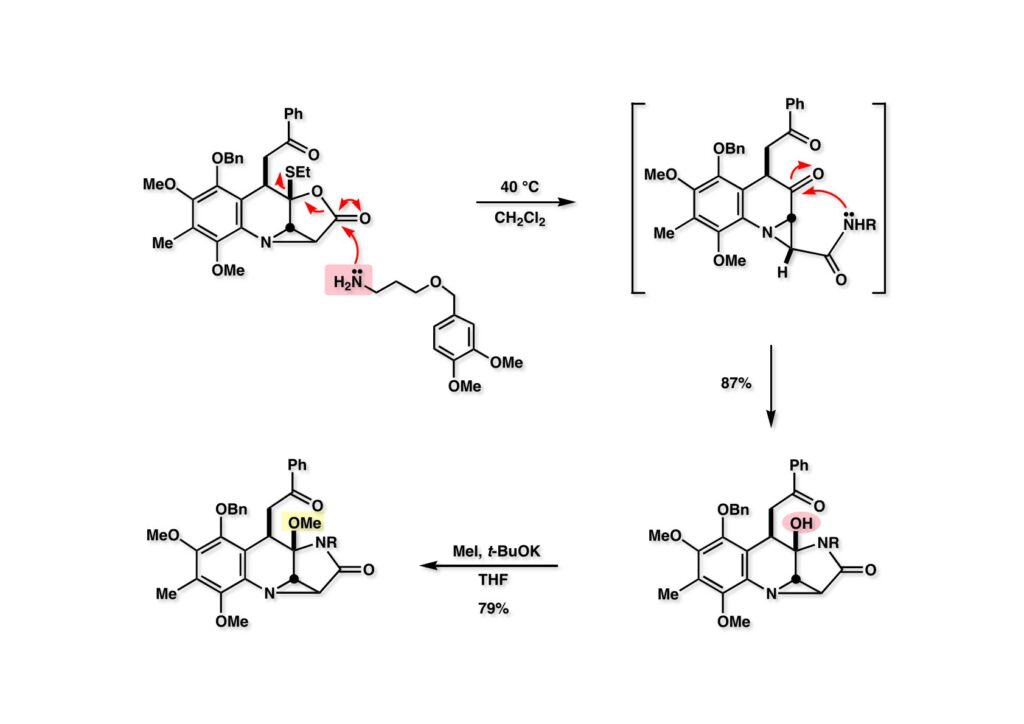 ラクトン (1-1) の開環はCH2Cl2中で環流するくらいで容易に進行したが、合成後期にうまく脱保護できるアミンがなかなか見つからなかった。当然、ベンジルアミンを最初に用いたが、加水素分解できなかった。(1-2) の様な、保護基に保護基を付けたようなみっともないアミンを使わざるを得なかったのは、それなりの背景があったのだ。アミノリシスで生じたアミド (1-3) はケトンを攻撃して、ヒドロキシラクタム (2-2) を与えた。次いで、比較的酸性度の大きい水酸基はMeI-t-BuOKの条件でメチル化されて (2-1) が得られた。
ラクトン (1-1) の開環はCH2Cl2中で環流するくらいで容易に進行したが、合成後期にうまく脱保護できるアミンがなかなか見つからなかった。当然、ベンジルアミンを最初に用いたが、加水素分解できなかった。(1-2) の様な、保護基に保護基を付けたようなみっともないアミンを使わざるを得なかったのは、それなりの背景があったのだ。アミノリシスで生じたアミド (1-3) はケトンを攻撃して、ヒドロキシラクタム (2-2) を与えた。次いで、比較的酸性度の大きい水酸基はMeI-t-BuOKの条件でメチル化されて (2-1) が得られた。
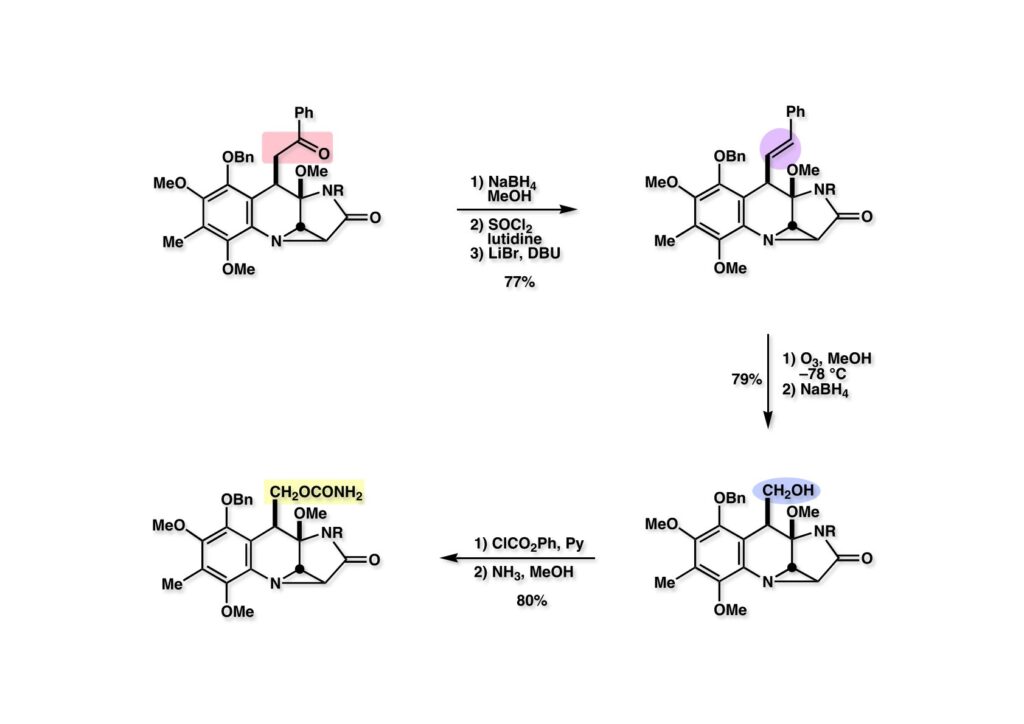 さて、次の仕事は全合成前期に重要な役割を果たしたカルコンの残骸であるフェナシル基をヒドロキシメチル基に変換することである。ケトン (1-1) を還元して、水酸基を塩化チオニルで脱水しようとしたが、クロライドになっただけだった。そこで LiBr存在下にDBUで加熱したところオレフィン (1-2) が得られた。オレフィン (1-2) にメタノール中低温でオゾンを通じたのち、NaBH4を加えてヒドロキシメチル体 (2-2) を得た。次いで、PhOCOCl-Pyで混合炭酸エステルとし、これを加アンモニア分解してカルバメート (2-1) に変換した。
さて、次の仕事は全合成前期に重要な役割を果たしたカルコンの残骸であるフェナシル基をヒドロキシメチル基に変換することである。ケトン (1-1) を還元して、水酸基を塩化チオニルで脱水しようとしたが、クロライドになっただけだった。そこで LiBr存在下にDBUで加熱したところオレフィン (1-2) が得られた。オレフィン (1-2) にメタノール中低温でオゾンを通じたのち、NaBH4を加えてヒドロキシメチル体 (2-2) を得た。次いで、PhOCOCl-Pyで混合炭酸エステルとし、これを加アンモニア分解してカルバメート (2-1) に変換した。
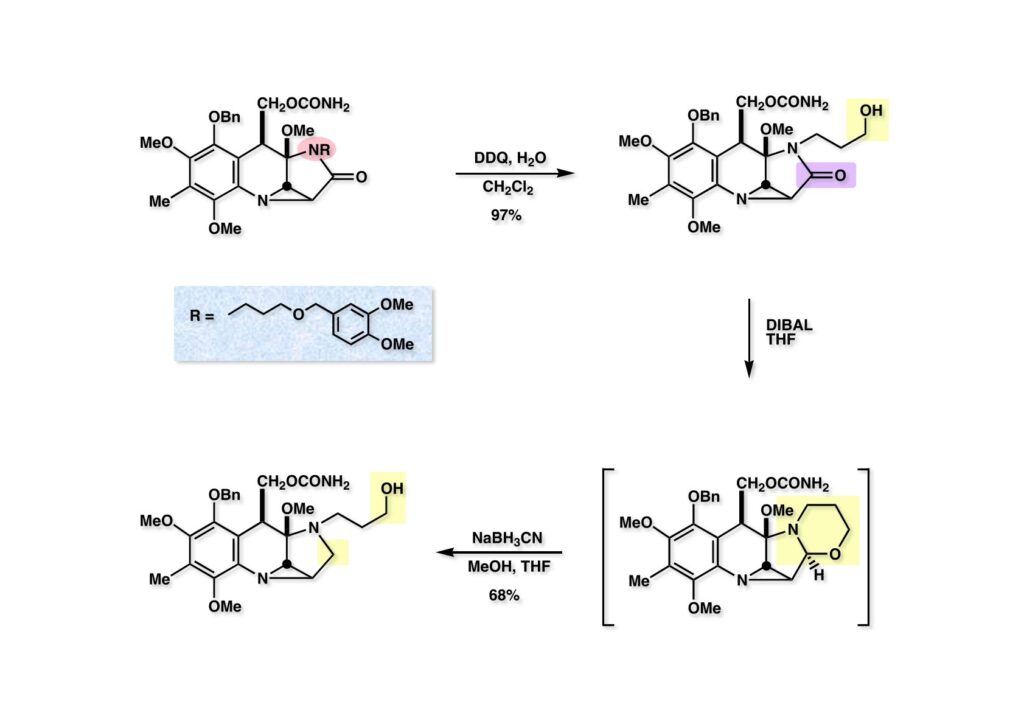 3,4-Dimethoxybenzyl基は北大薬の及川、米光により報告されたアルコールの保護機で、DDQ酸化により容易に除去できる。(1-1) の保護基の保護基を外して (1-2) をまず得た。これをDIBAL還元すると、通常のN-アルキル基なら直接アミンにまで還元されるが、水酸基が存在するので1,3-oxazinane (2-2) で反応が止まってしまう。仕方ないので、反応溶液にメタノールとNaBH3CNを加えてアミン (2-1) を得た。
3,4-Dimethoxybenzyl基は北大薬の及川、米光により報告されたアルコールの保護機で、DDQ酸化により容易に除去できる。(1-1) の保護基の保護基を外して (1-2) をまず得た。これをDIBAL還元すると、通常のN-アルキル基なら直接アミンにまで還元されるが、水酸基が存在するので1,3-oxazinane (2-2) で反応が止まってしまう。仕方ないので、反応溶液にメタノールとNaBH3CNを加えてアミン (2-1) を得た。
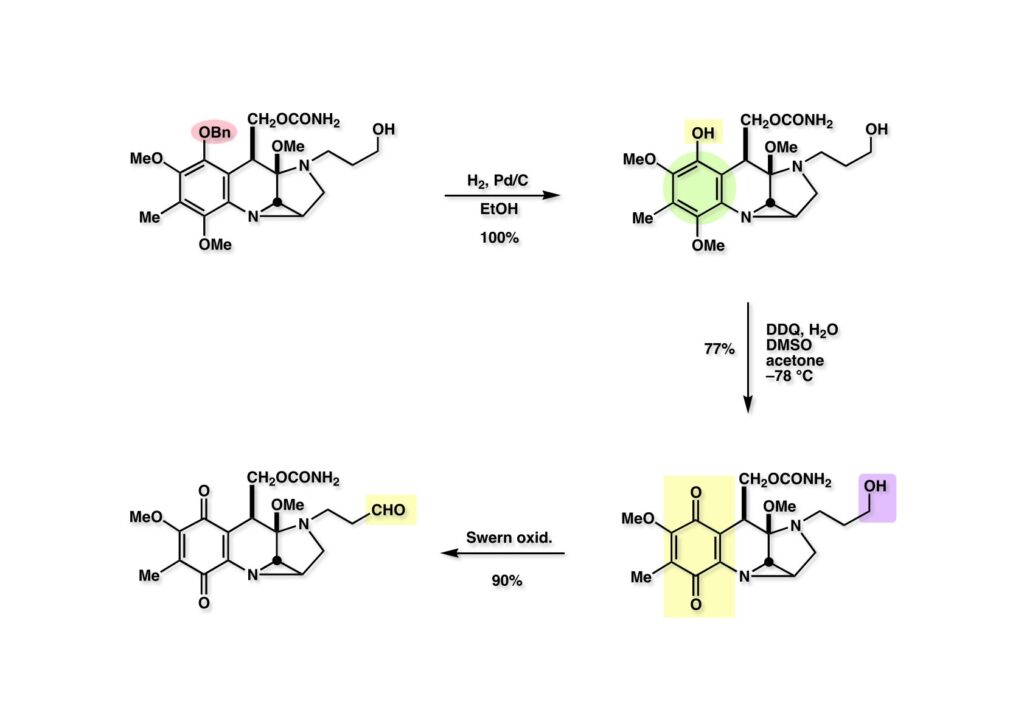 次に芳香環を酸化してキノンに変換するステージがやってきた。(1-1) のベンジルエーテルは接触還元で容易に除去でき、フェノール (1-2) が定量的に得られた。イソマイトマイシンはマイトマイシンに比べると酸性条件に弱いと記述したが、(1-2) のDDQ酸化の条件でも常温で反応させると分解が部分的に起こる。そのため、低温下で酸化する必要があるが、(1-2) がアセトンに溶けにくかったのでDMSOを補助溶媒に用いたところ、p-キノン/o-キノンの比が改善され、(2-2) の収率も向上した。(注)DDQは酸化することによりジフェノールになるが環上にシアノ基やクロル基などの電子吸引基が存在しているため、フェノールよりは酸性度の大きい酸と言える。
次に芳香環を酸化してキノンに変換するステージがやってきた。(1-1) のベンジルエーテルは接触還元で容易に除去でき、フェノール (1-2) が定量的に得られた。イソマイトマイシンはマイトマイシンに比べると酸性条件に弱いと記述したが、(1-2) のDDQ酸化の条件でも常温で反応させると分解が部分的に起こる。そのため、低温下で酸化する必要があるが、(1-2) がアセトンに溶けにくかったのでDMSOを補助溶媒に用いたところ、p-キノン/o-キノンの比が改善され、(2-2) の収率も向上した。(注)DDQは酸化することによりジフェノールになるが環上にシアノ基やクロル基などの電子吸引基が存在しているため、フェノールよりは酸性度の大きい酸と言える。
 岸研でMMCを合成したときはアジリジンに結合した-CH2CH2CHOをPhNMe2-HClO4という弱酸条件でretro-Michael反応で除去した。ところが、この条件ですらIMMAは分解しがちで、副生物やMMAへの転位反応も観測された。より穏和な条件を模索して、エナミン (1-2) を構築するような条件にすれば良いかもしれないと思った。(1-1) を5当量のピロリジンと10当量の酢酸でCH2Cl2室温で1.5時間放置したところ、70%の収率でイソマイトマイシンA (2-1) が得られた。
岸研でMMCを合成したときはアジリジンに結合した-CH2CH2CHOをPhNMe2-HClO4という弱酸条件でretro-Michael反応で除去した。ところが、この条件ですらIMMAは分解しがちで、副生物やMMAへの転位反応も観測された。より穏和な条件を模索して、エナミン (1-2) を構築するような条件にすれば良いかもしれないと思った。(1-1) を5当量のピロリジンと10当量の酢酸でCH2Cl2室温で1.5時間放置したところ、70%の収率でイソマイトマイシンA (2-1) が得られた。
 マイトマイシン転位は協和発酵の報告どおり、IMMA (1-1) を1当量のAl(OPr-i)3存在下でメタノール中室温で4日間放置することで、MMA (1-2)を高収率で得ることができた。最後にMMA (1-2) のメタノール溶液にアンモニアのメタノール溶液を加えることでマイトマイシンC (2-1) の全合成が完成した。大学院1年生のLihu Yangを毎日突っつきながら、全合成開始後1年と数日でMMCの全合成は終わったが、まだまだ改良したい点があって、直ちに第二世代の全合成に着手するようにLihuに伝えた。
マイトマイシン転位は協和発酵の報告どおり、IMMA (1-1) を1当量のAl(OPr-i)3存在下でメタノール中室温で4日間放置することで、MMA (1-2)を高収率で得ることができた。最後にMMA (1-2) のメタノール溶液にアンモニアのメタノール溶液を加えることでマイトマイシンC (2-1) の全合成が完成した。大学院1年生のLihu Yangを毎日突っつきながら、全合成開始後1年と数日でMMCの全合成は終わったが、まだまだ改良したい点があって、直ちに第二世代の全合成に着手するようにLihuに伝えた。
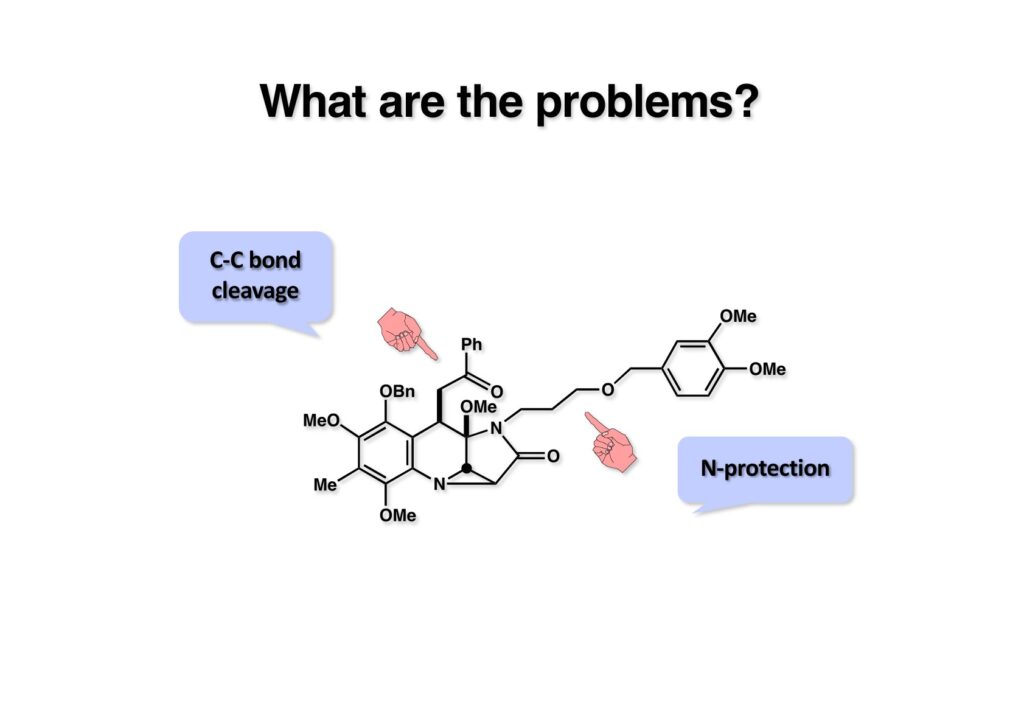 何が君は不満なんだい?と聞かれれば、合成初期のカルコンへのMichael付加で得られたシリルエノールエーテルの二重結合を開裂に使えなかったことと、保護基の保護基というダサいアミンを使わざるを得なかったことである。少なくともこの2点を改良すべく次の全合成ルートを考えて実行することにした。
何が君は不満なんだい?と聞かれれば、合成初期のカルコンへのMichael付加で得られたシリルエノールエーテルの二重結合を開裂に使えなかったことと、保護基の保護基というダサいアミンを使わざるを得なかったことである。少なくともこの2点を改良すべく次の全合成ルートを考えて実行することにした。
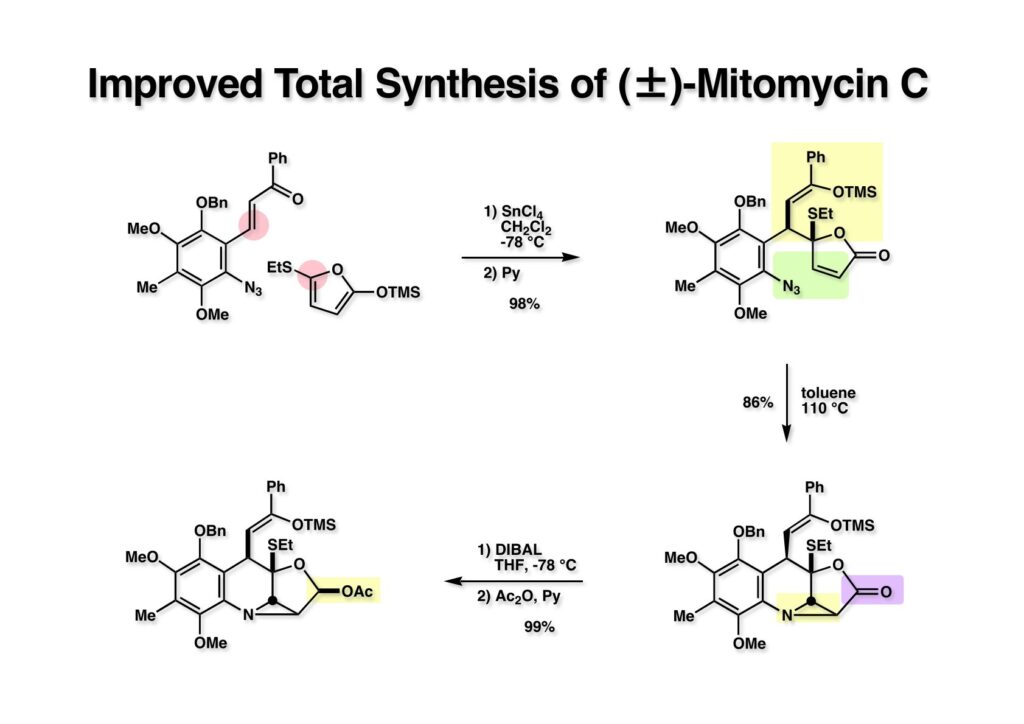 カルコン (1-1) へのフラン体 (1-2) のMichael付加は第一世代合成と同様であるが、後処理中にシリルエノールエーテルの加水分解を防ぐためにピリジンを加えてから後処理して付加体 (1-3) を得た。次にトルエン中還流してアジリジン (2-2) に導いた。ここでラクトン (2-2) をDIBAL還元し、生じたラクトールをアセチル化することでアセテート (2-1) に導いた。ところで、このアセテートの立体化学であるがconvex面から反応が起こるのが当然と思っていたので第二世代の論文にもアセトキシ基の立体化学はα配置と描いた。その後に光学活性体のMMCの合成をMaria Tomasz教授から依頼されたので、ラクトールをマンデル酸でエステル化してX線解析をしたところβ配置になっており、驚いてしまった。この事実は第二世代合成の成功に関わる重要な現象なので次ページで説明したい。
カルコン (1-1) へのフラン体 (1-2) のMichael付加は第一世代合成と同様であるが、後処理中にシリルエノールエーテルの加水分解を防ぐためにピリジンを加えてから後処理して付加体 (1-3) を得た。次にトルエン中還流してアジリジン (2-2) に導いた。ここでラクトン (2-2) をDIBAL還元し、生じたラクトールをアセチル化することでアセテート (2-1) に導いた。ところで、このアセテートの立体化学であるがconvex面から反応が起こるのが当然と思っていたので第二世代の論文にもアセトキシ基の立体化学はα配置と描いた。その後に光学活性体のMMCの合成をMaria Tomasz教授から依頼されたので、ラクトールをマンデル酸でエステル化してX線解析をしたところβ配置になっており、驚いてしまった。この事実は第二世代合成の成功に関わる重要な現象なので次ページで説明したい。
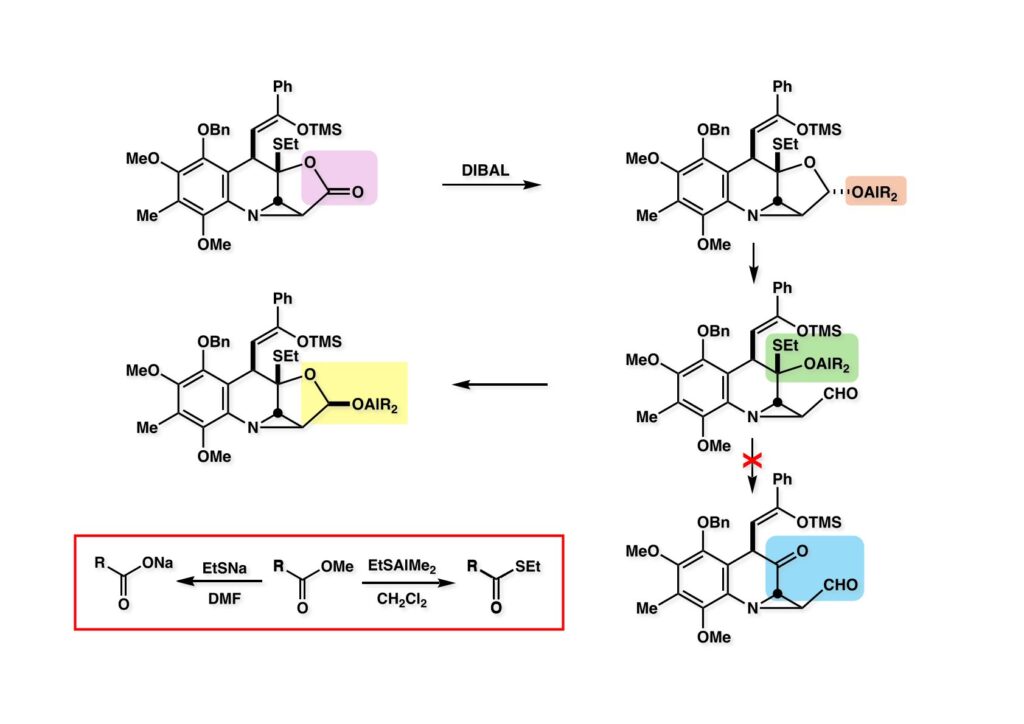 ラクトン (1-1) をDIBALで還元すると、お椀型の分子なので試薬は十中八九convex面から攻撃すると考えられるので (1-2) が生成するだろう。おそらく可逆的な反応でラクトールの開環が起きるとすると (2-2) が生成する。ここでもしR2AlSEtが脱離すればケトアルデヒド (3-4) に変換され、DIBALによってさらに還元されると考えられ、ゴチャゴチャいじれば何とかなっただろうが、苦労することは目に見えている。ところが実際は、(2-2) でアルコキシド (2-2) は直ちに閉環して (1-2) より安定なラクトール (2-2) を安定な化合物として与えた。この経路は結合エネルギーに関係しているだろう。即ち、bond dissociation energyの表をもとにして乱暴な議論をするとすれば、Al-S (89.4 kcal/mol)、Al-O (122.4 kcal/mol) で、(2-2) においてEtSAlR2を追い出す方がエネルギー的に不利であり、環化しなおしてAl-O結合を生成した方が有利となる理屈だ。ちなみにメチルエステル (3-2) もEtSAlR2と反応させるとチオエステル (3-3) に変換できるがNa塩ではそうはいかない。DMF中でEtSNaと加熱すると脱メチル化が起きるだけである。
ラクトン (1-1) をDIBALで還元すると、お椀型の分子なので試薬は十中八九convex面から攻撃すると考えられるので (1-2) が生成するだろう。おそらく可逆的な反応でラクトールの開環が起きるとすると (2-2) が生成する。ここでもしR2AlSEtが脱離すればケトアルデヒド (3-4) に変換され、DIBALによってさらに還元されると考えられ、ゴチャゴチャいじれば何とかなっただろうが、苦労することは目に見えている。ところが実際は、(2-2) でアルコキシド (2-2) は直ちに閉環して (1-2) より安定なラクトール (2-2) を安定な化合物として与えた。この経路は結合エネルギーに関係しているだろう。即ち、bond dissociation energyの表をもとにして乱暴な議論をするとすれば、Al-S (89.4 kcal/mol)、Al-O (122.4 kcal/mol) で、(2-2) においてEtSAlR2を追い出す方がエネルギー的に不利であり、環化しなおしてAl-O結合を生成した方が有利となる理屈だ。ちなみにメチルエステル (3-2) もEtSAlR2と反応させるとチオエステル (3-3) に変換できるがNa塩ではそうはいかない。DMF中でEtSNaと加熱すると脱メチル化が起きるだけである。
 シリルエノールエーテル (1-1) のオゾン分解はきれいな結果を出さなかったのでRuO4酸化を試みた。この酸化にはRuCl3-NaIO4を使う方が多いが、私はRuO2-NaIO4を以前から使っていた。ここで注意しなければならないのはRuO2・nH2Oを使うことで、無水のRuO2を使うと全く溶解せずに無反応で終わる。RuO4はおとなしそうな試薬だが、芳香環の開裂、アミドやウレタンの結合した炭素の酸化、アルコールをカルボン酸にまで酸化などなど、結構大胆な反応をやってくれる私の「お気に入り」である。ここでは、NaIO4の1当量目でスルフィドがスルフォキサイドに、次の1当量でスルフォンに、そして3当量目でシリルエノールエーテルが開裂してアルデヒド (1-2) が生成する。通常はアルデヒドがさらに酸化されてカルボン酸になってしまうが、ここでは立体障害のためかきれいにアルデヒド (1-2) が単離される。アルデヒド (1-2) をNaBH4還元してアルコール (2-2) に変換し、さらにtrichloroacetyl isocyanateと反応させて保護されたカルバメート (2-1) を得た。
シリルエノールエーテル (1-1) のオゾン分解はきれいな結果を出さなかったのでRuO4酸化を試みた。この酸化にはRuCl3-NaIO4を使う方が多いが、私はRuO2-NaIO4を以前から使っていた。ここで注意しなければならないのはRuO2・nH2Oを使うことで、無水のRuO2を使うと全く溶解せずに無反応で終わる。RuO4はおとなしそうな試薬だが、芳香環の開裂、アミドやウレタンの結合した炭素の酸化、アルコールをカルボン酸にまで酸化などなど、結構大胆な反応をやってくれる私の「お気に入り」である。ここでは、NaIO4の1当量目でスルフィドがスルフォキサイドに、次の1当量でスルフォンに、そして3当量目でシリルエノールエーテルが開裂してアルデヒド (1-2) が生成する。通常はアルデヒドがさらに酸化されてカルボン酸になってしまうが、ここでは立体障害のためかきれいにアルデヒド (1-2) が単離される。アルデヒド (1-2) をNaBH4還元してアルコール (2-2) に変換し、さらにtrichloroacetyl isocyanateと反応させて保護されたカルバメート (2-1) を得た。
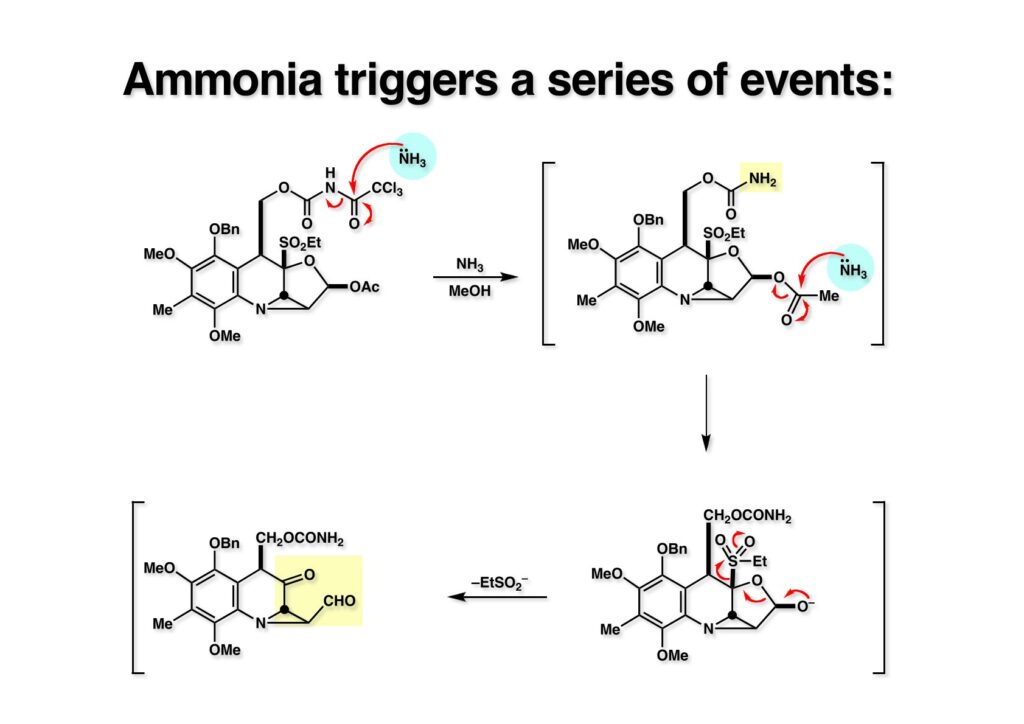 さて、次のスキームは流れるように進行して、私の計画通りだと自己満足している。(1-1) をメタノール中アンモニアで処理すると、まずトリクロロアセチル基が脱落する。次に (1-2) のアセチル基が加アンモニア分解されてラクトール (2-2) が生成するがEtSO2- (ehtyl sulfinate)が効果的な脱離基となってケトアルデヒド (2-1) になる。
さて、次のスキームは流れるように進行して、私の計画通りだと自己満足している。(1-1) をメタノール中アンモニアで処理すると、まずトリクロロアセチル基が脱落する。次に (1-2) のアセチル基が加アンモニア分解されてラクトール (2-2) が生成するがEtSO2- (ehtyl sulfinate)が効果的な脱離基となってケトアルデヒド (2-1) になる。
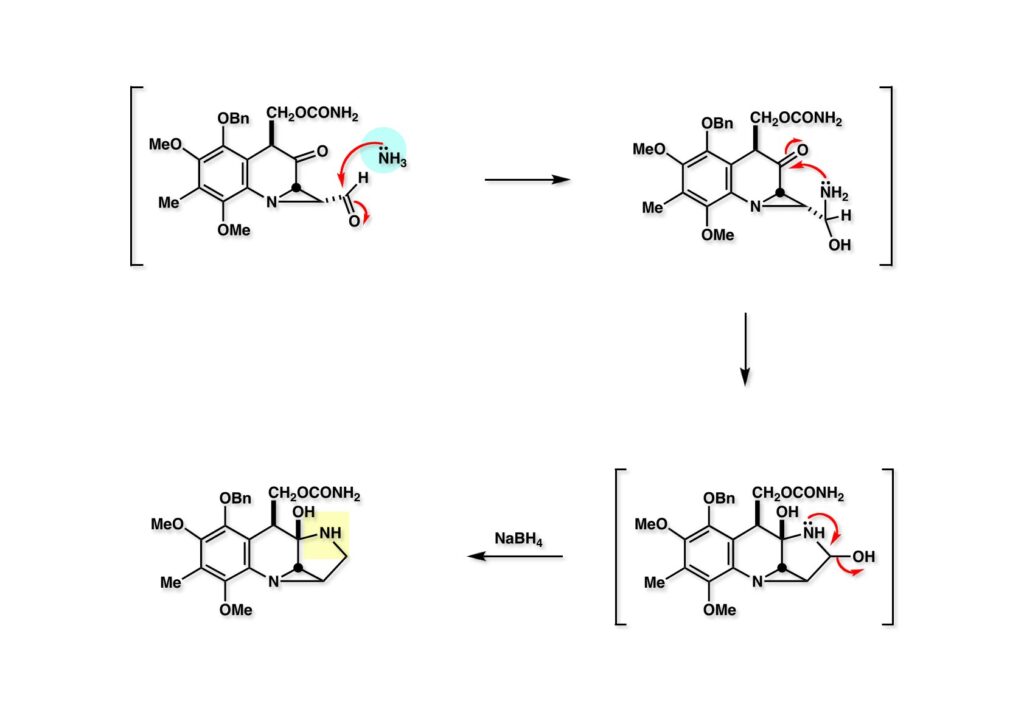 次いでアンモニアがアルデヒド (1-1) に付加してヘミアミナール (1-2) となり、更にケトンと反応してビスヘミアミナール (2-2) に変換されたはずである。実はこの反応を初めてやった時に、Lihuが私のオフィスに泣きそうな顔で入ってきて、「TLCをチェックしたらスポットが消えてしまった」と泣き言を言った。私は「ばか、この化合物がTLC上でまともに見えるはずがないから、サッサとNaBH4を放り込め」と追い返した、少し経ってLihuが満面に笑みを浮かべて (2-1) のスポットが現れたと報告した。残ったヘミアミナールは橋頭位に位置するので簡単に還元される訳はないと思っていたが正解であった。ちなみに、カルバメートに変換する前のアルコール体からヘミアミナール (2-1) への収率は61%であった。
次いでアンモニアがアルデヒド (1-1) に付加してヘミアミナール (1-2) となり、更にケトンと反応してビスヘミアミナール (2-2) に変換されたはずである。実はこの反応を初めてやった時に、Lihuが私のオフィスに泣きそうな顔で入ってきて、「TLCをチェックしたらスポットが消えてしまった」と泣き言を言った。私は「ばか、この化合物がTLC上でまともに見えるはずがないから、サッサとNaBH4を放り込め」と追い返した、少し経ってLihuが満面に笑みを浮かべて (2-1) のスポットが現れたと報告した。残ったヘミアミナールは橋頭位に位置するので簡単に還元される訳はないと思っていたが正解であった。ちなみに、カルバメートに変換する前のアルコール体からヘミアミナール (2-1) への収率は61%であった。
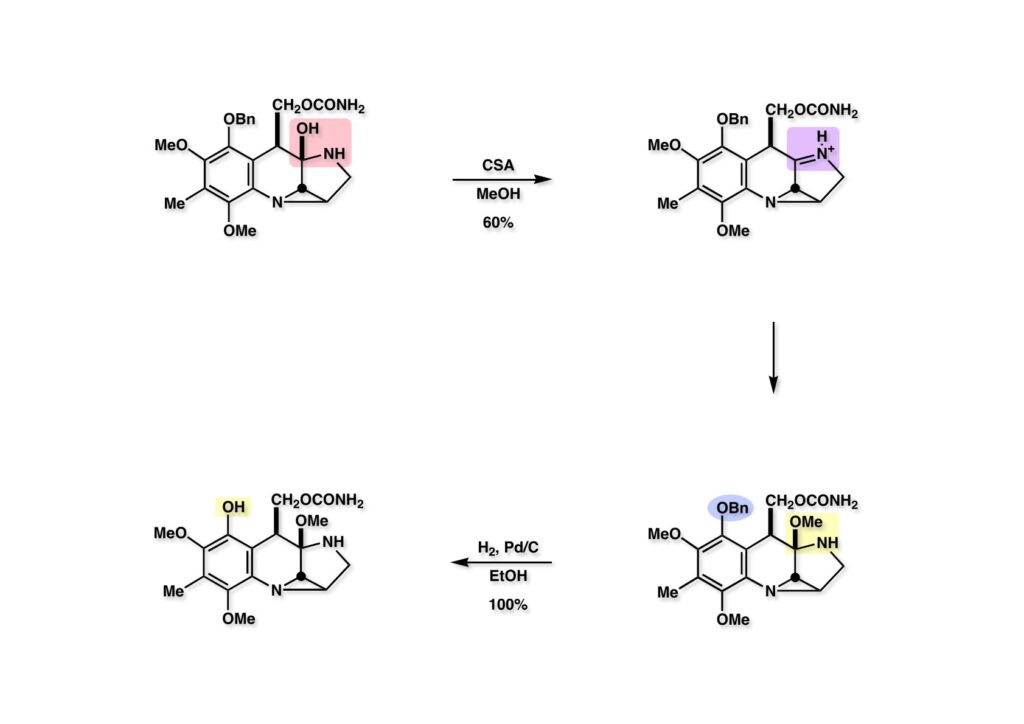 ヘミアミナール (1-1) のメチル化は橋頭位ながらもひょっとしたらイミニウム塩 (1-2) が生成するかもしれないと、メタノール中で触媒量のCSAを加えて室温放置したところ、首尾よくメタノールが付加した化合物 (2-2) が得られた。これ以後に問題となるステップは予見できなかったので気楽に合成を進めた。(2-2) のベンジル基を加水素分解してフェノール (2-1) に変換した。
ヘミアミナール (1-1) のメチル化は橋頭位ながらもひょっとしたらイミニウム塩 (1-2) が生成するかもしれないと、メタノール中で触媒量のCSAを加えて室温放置したところ、首尾よくメタノールが付加した化合物 (2-2) が得られた。これ以後に問題となるステップは予見できなかったので気楽に合成を進めた。(2-2) のベンジル基を加水素分解してフェノール (2-1) に変換した。
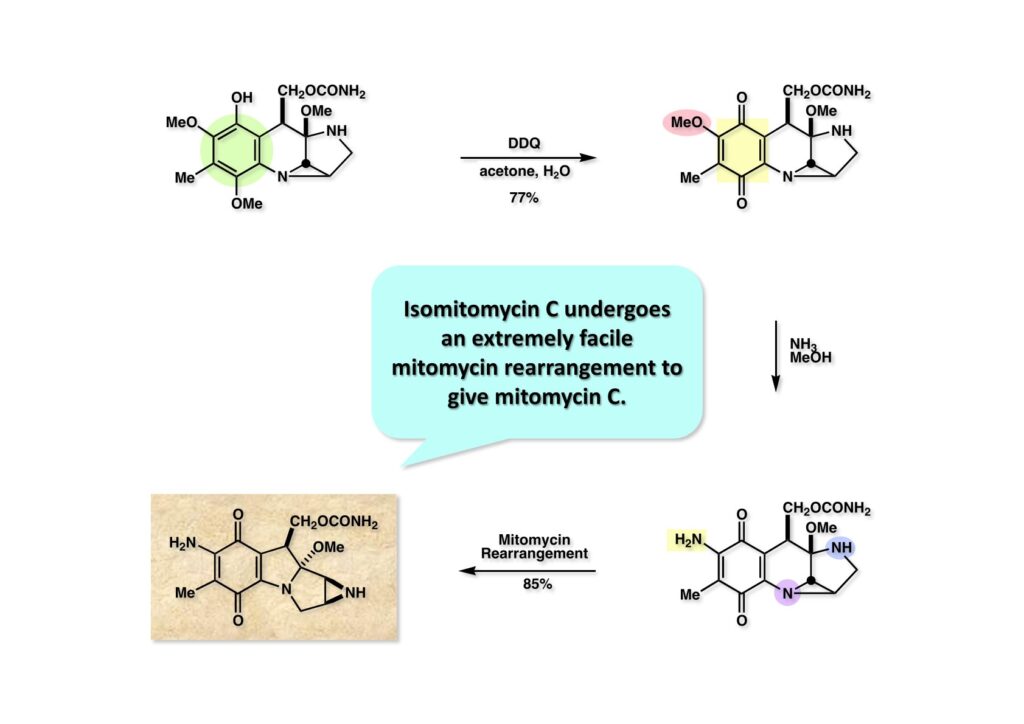 フェノール (1-1) を低温下DDQ酸化してイソマイトマイシンA (1-2) を得た。以前、協和発酵の担当者にイソマイトマイシンCを見たことがあるか尋ねたところ、それらしき化合物があると思ってTLCで分離しようと思ったらマイトマイシンCになっていた、との返事があった。これはマイトマイシン転位が非常に早いのではと思い、イソマイトマイシンA (1-2) をメタノールに溶かしてアンモニアを加え、数時間室温で放置したところマイトマイシンC (2-1) が高収率で得られた。イソマイトマイシンC (2-2) は幻の化合物でその存在を認めるには至らなかった。マイトマイシンCの全合成は1年半で完成し、それほど必死に努力したわけでもなかったのに高評価が得られた。この世界、苦労すれば苦労しただけ報われる、っていうわけでもないね。
フェノール (1-1) を低温下DDQ酸化してイソマイトマイシンA (1-2) を得た。以前、協和発酵の担当者にイソマイトマイシンCを見たことがあるか尋ねたところ、それらしき化合物があると思ってTLCで分離しようと思ったらマイトマイシンCになっていた、との返事があった。これはマイトマイシン転位が非常に早いのではと思い、イソマイトマイシンA (1-2) をメタノールに溶かしてアンモニアを加え、数時間室温で放置したところマイトマイシンC (2-1) が高収率で得られた。イソマイトマイシンC (2-2) は幻の化合物でその存在を認めるには至らなかった。マイトマイシンCの全合成は1年半で完成し、それほど必死に努力したわけでもなかったのに高評価が得られた。この世界、苦労すれば苦労しただけ報われる、っていうわけでもないね。